

EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing
- PMID: 24893890
- PMCID: PMC4125473
- DOI: 10.1158/2159-8290.CD-13-0879
EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing
Abstract
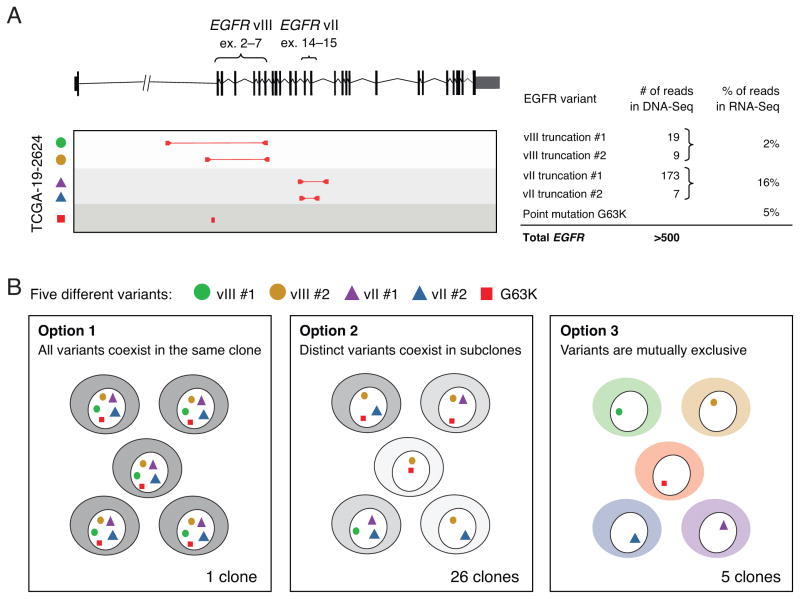
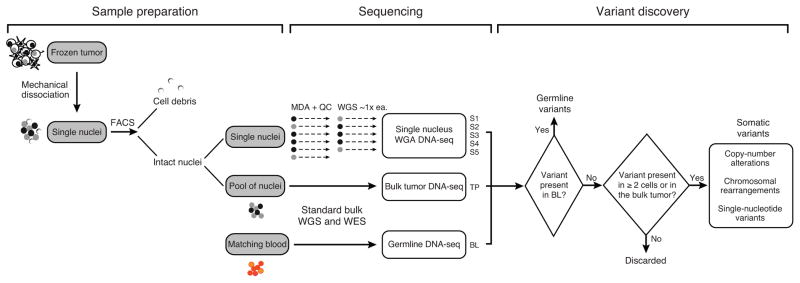
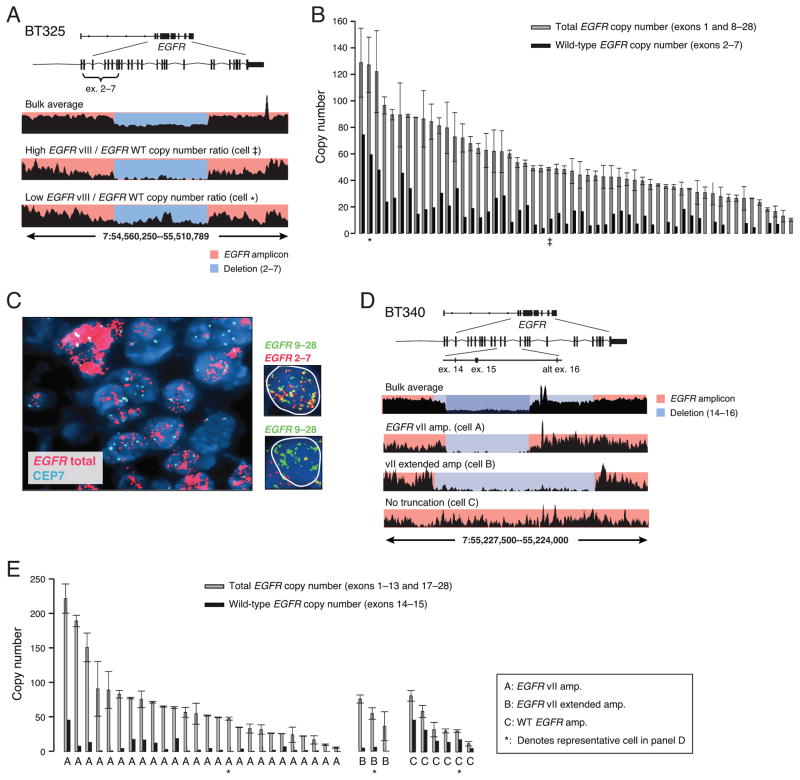
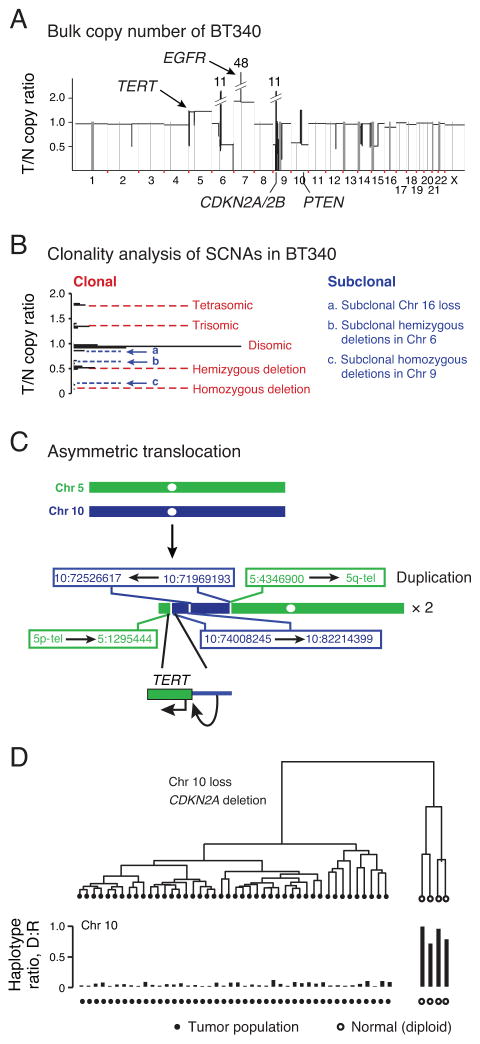
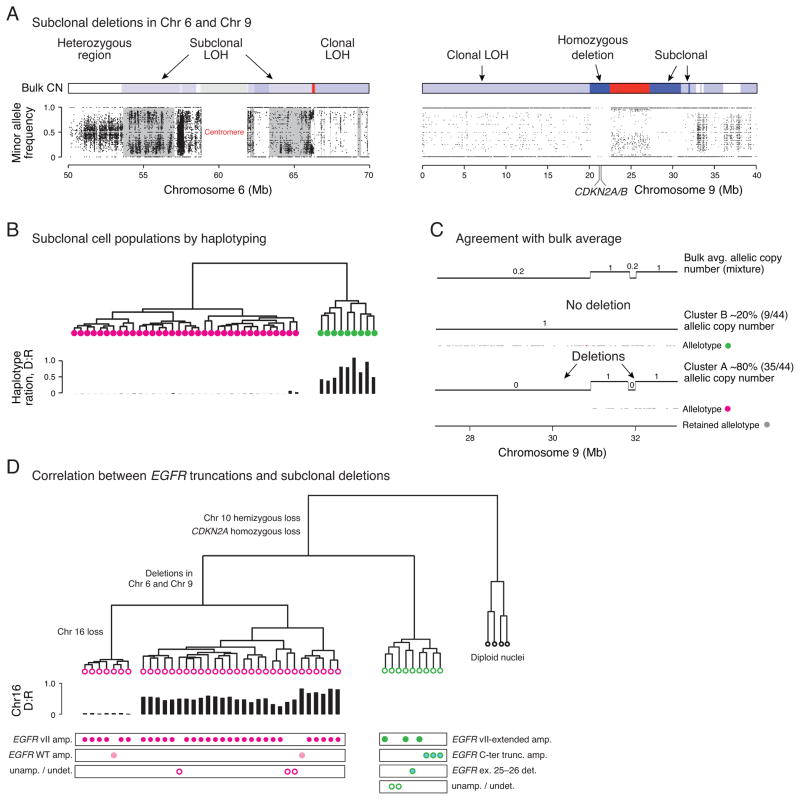
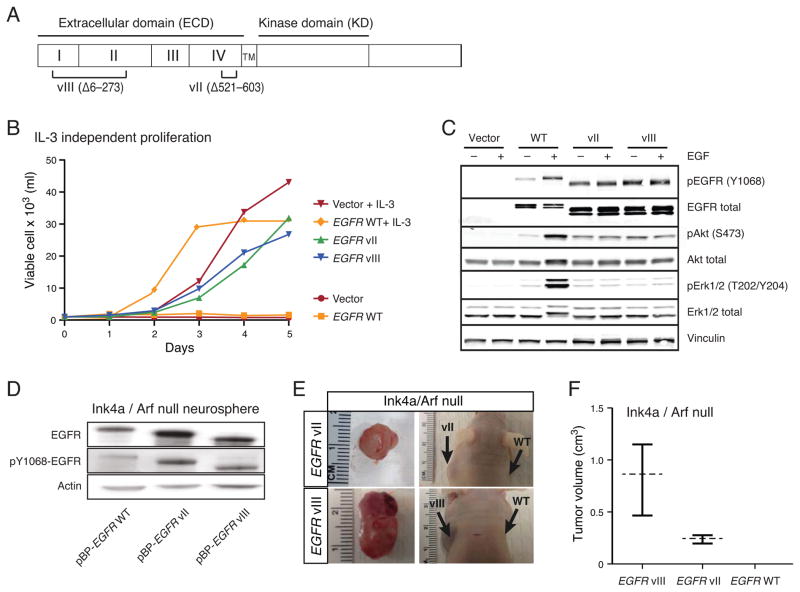
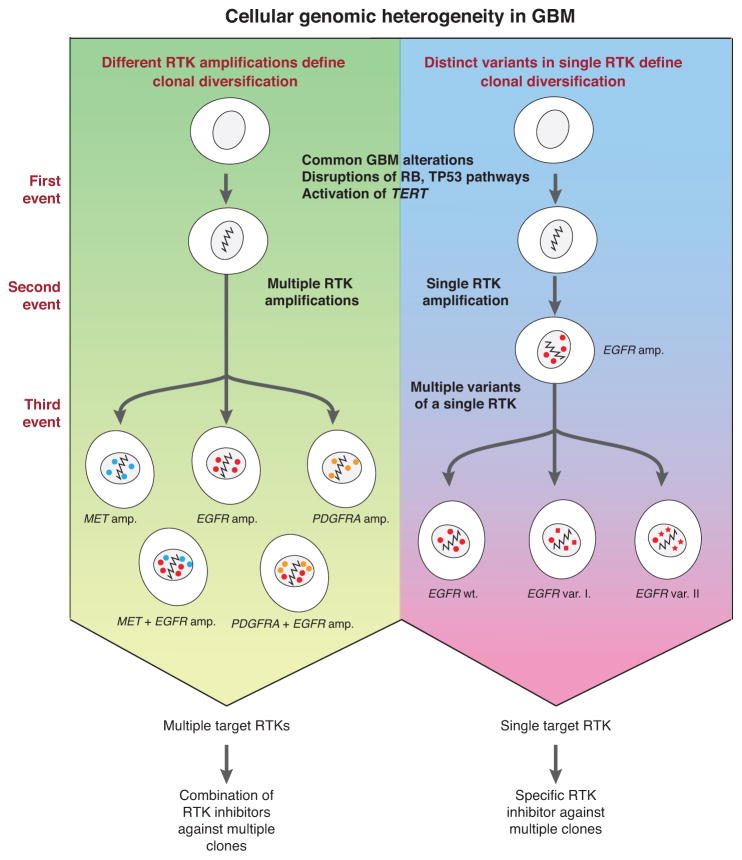
Glioblastomas (GBM) with EGFR amplification represent approximately 50% of newly diagnosed cases, and recent studies have revealed frequent coexistence of multiple EGFR aberrations within the same tumor, which has implications for mutation cooperation and treatment resistance. However, bulk tumor sequencing studies cannot resolve the patterns of how the multiple EGFR aberrations coexist with other mutations within single tumor cells. Here, we applied a population-based single-cell whole-genome sequencing methodology to characterize genomic heterogeneity in EGFR-amplified glioblastomas. Our analysis effectively identified clonal events, including a novel translocation of a super enhancer to the TERT promoter, as well as subclonal LOH and multiple EGFR mutational variants within tumors. Correlating the EGFR mutations onto the cellular hierarchy revealed that EGFR truncation variants (EGFRvII and EGFR carboxyl-terminal deletions) identified in the bulk tumor segregate into nonoverlapping subclonal populations. In vitro and in vivo functional studies show that EGFRvII is oncogenic and sensitive to EGFR inhibitors currently in clinical trials. Thus, the association between diverse activating mutations in EGFR and other subclonal mutations within a single tumor supports an intrinsic mechanism for proliferative and clonal diversification with broad implications in resistance to treatment.
Significance: We developed a novel single-cell sequencing methodology capable of identifying unique, nonoverlapping subclonal alterations from archived frozen clinical specimens. Using GBM as an example, we validated our method to successfully define tumor cell subpopulations containing distinct genetic and treatment resistance profiles and potentially mutually cooperative combinations of alterations in EGFR and other genes.
©2014 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment in
-
Greater than the sum of its parts: single-nucleus sequencing identifies convergent evolution of independent EGFR mutants in GBM.Cancer Discov. 2014 Aug;4(8):876-8. doi: 10.1158/2159-8290.CD-14-0635. Cancer Discov. 2014. PMID: 25092745 Free PMC article.
References
-
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
