

Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND)
- PMID: 24894206
- PMCID: PMC4043700
- DOI: 10.1186/1742-6405-11-13
Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND)
Abstract
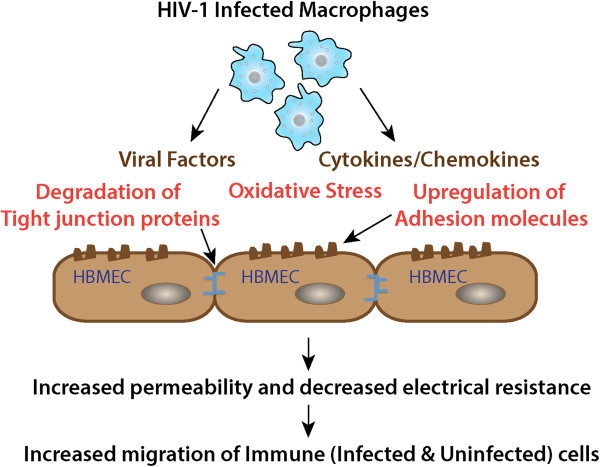
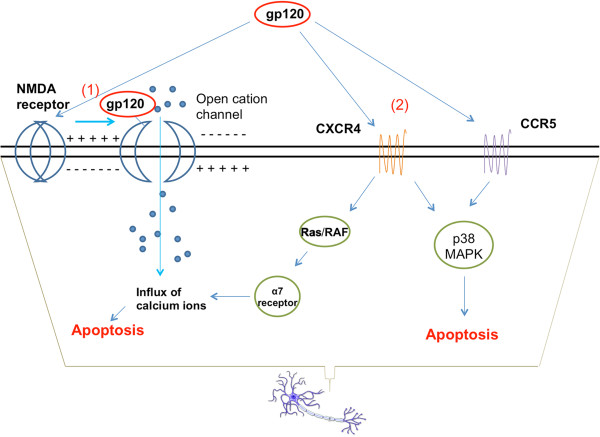
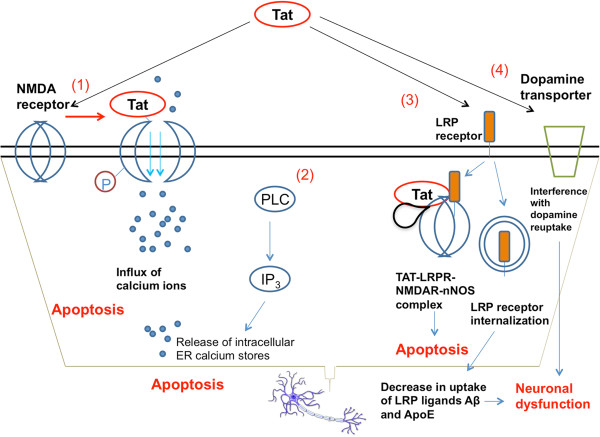
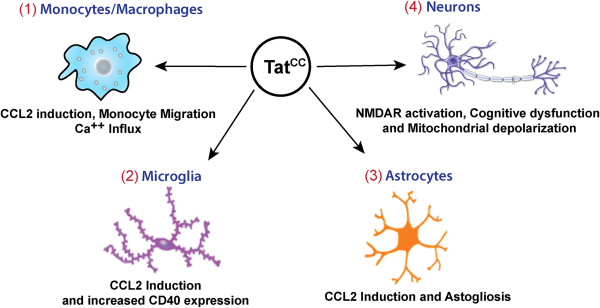
As the HIV-1 epidemic enters its fourth decade, HIV-1 associated neurological disorders (HAND) continue to be a major concern in the infected population, despite the widespread use of anti-retroviral therapy. Advancing age and increased life expectancy of the HIV-1 infected population have been shown to increase the risk of cognitive dysfunction. Over the past 10 years, there has been a significant progress in our understanding of the mechanisms and the risk factors involved in the development of HAND. Key events that lead up to neuronal damage in HIV-1 infected individuals can be categorized based on the interaction of HIV-1 with the various cell types, including but not limited to macrophages, brain endothelial cells, microglia, astrocytes and the neurons. This review attempts to decipher these interactions, beginning with HIV-1 infection of macrophages and ultimately resulting in the release of neurotoxic viral and host products. These include: interaction with endothelial cells, resulting in the impairment of the blood brain barrier; interaction with the astrocytes, leading to metabolic and neurotransmitter imbalance; interactions with resident immune cells in the brain, leading to release of toxic cytokines and chemokines. We also review the mechanisms underlying neuronal damage caused by the factors mentioned above. We have attempted to bring together recent findings in these areas to help appreciate the viral and host factors that bring about neurological dysfunction. In addition, we review host factors and viral genotypic differences that affect phenotypic pathological outcomes, as well as recent advances in treatment options to specifically address the neurotoxic mechanisms in play.
Figures
References
-
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, Group C, Group H. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;11(1):3–16. doi: 10.1007/s13365-010-0006-1. - DOI - PMC - PubMed
-
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;11(18):1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. - DOI - PMC - PubMed
-
- Airoldi M, Bandera A, Trabattoni D, Tagliabue B, Arosio B, Soria A, Rainone V, Lapadula G, Annoni G, Clerici M, Gori A. Neurocognitive impairment in HIV-infected naive patients with advanced disease: the role of virus and intrathecal immune activation. Clin Dev Immunol. 2012;11:467154. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
