

Bax inhibitor-1-mediated inhibition of mitochondrial Ca2+ intake regulates mitochondrial permeability transition pore opening and cell death
- PMID: 24899098
- PMCID: PMC4046133
- DOI: 10.1038/srep05194
Bax inhibitor-1-mediated inhibition of mitochondrial Ca2+ intake regulates mitochondrial permeability transition pore opening and cell death
Abstract
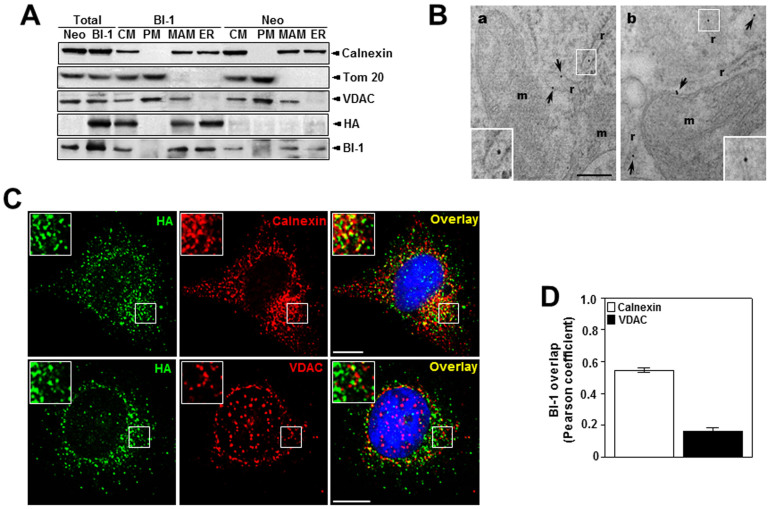
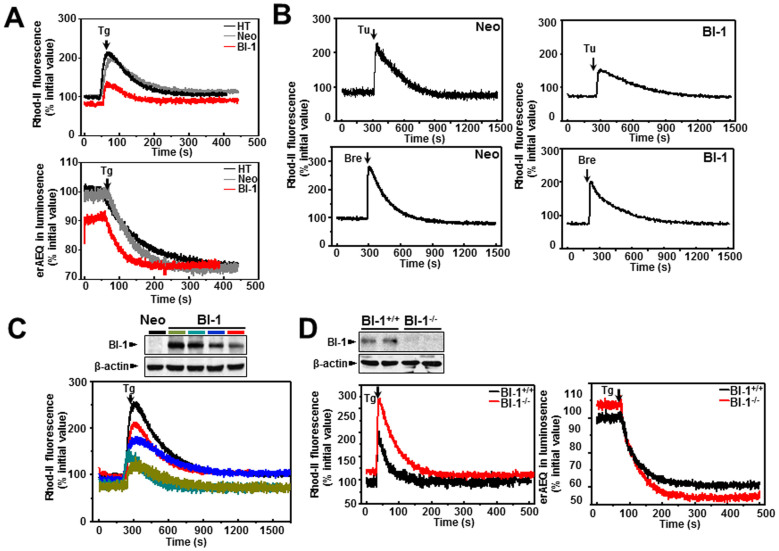
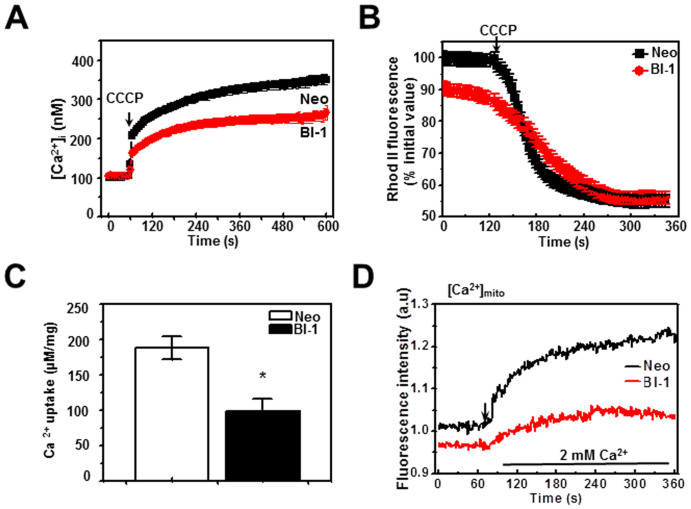
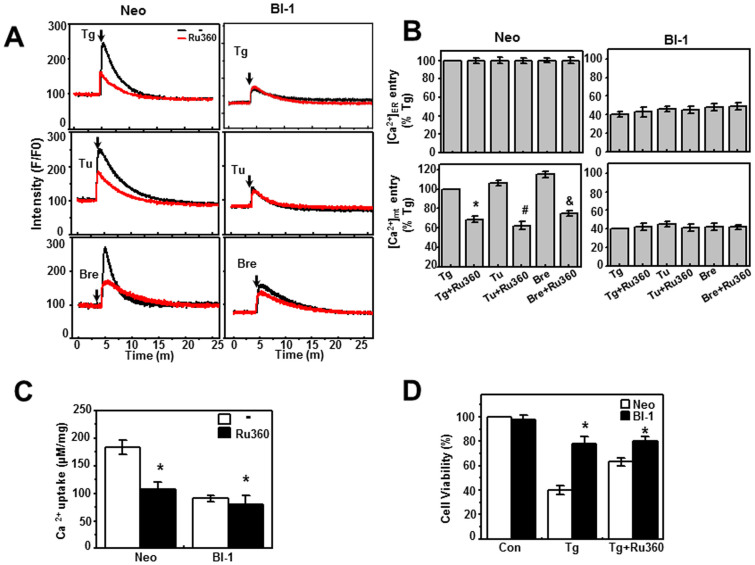
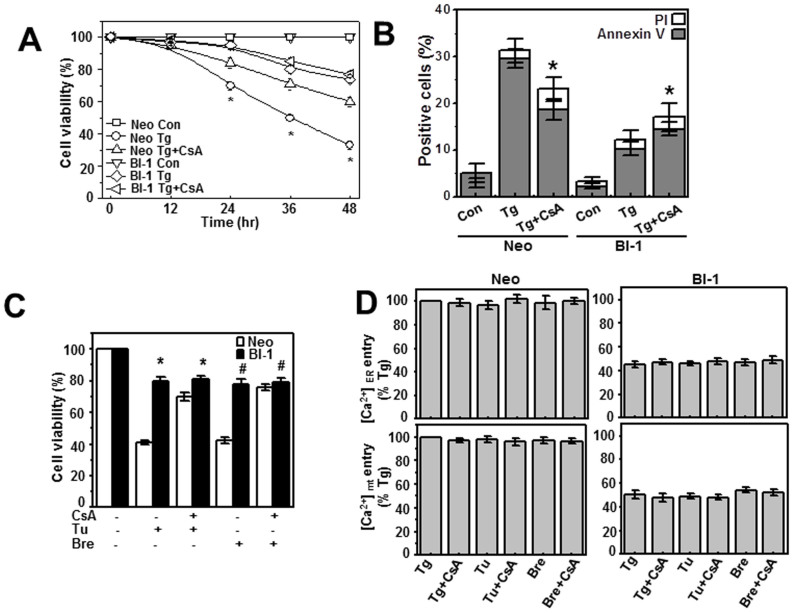
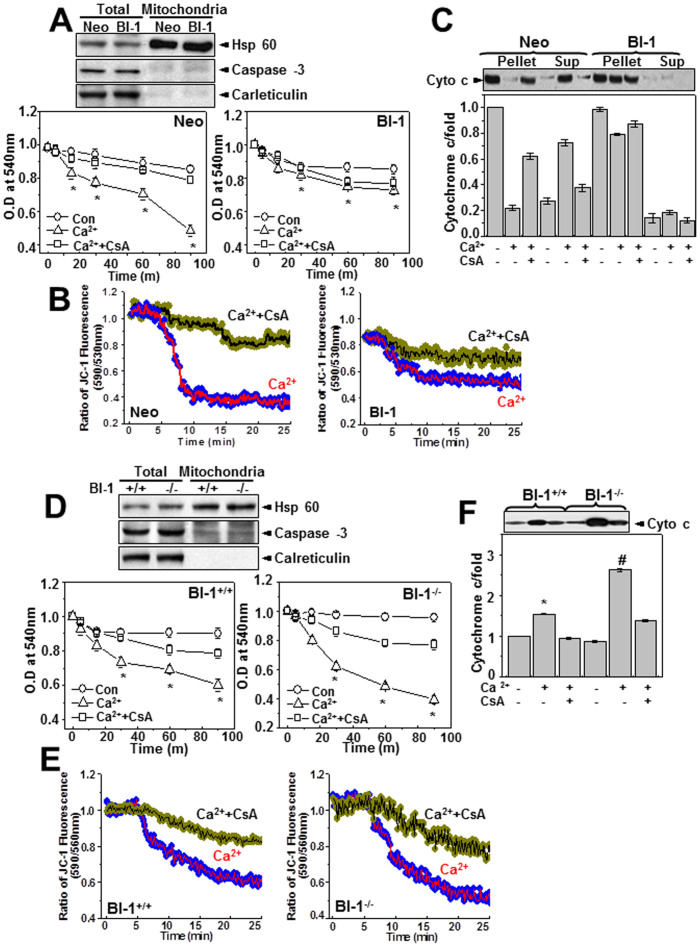
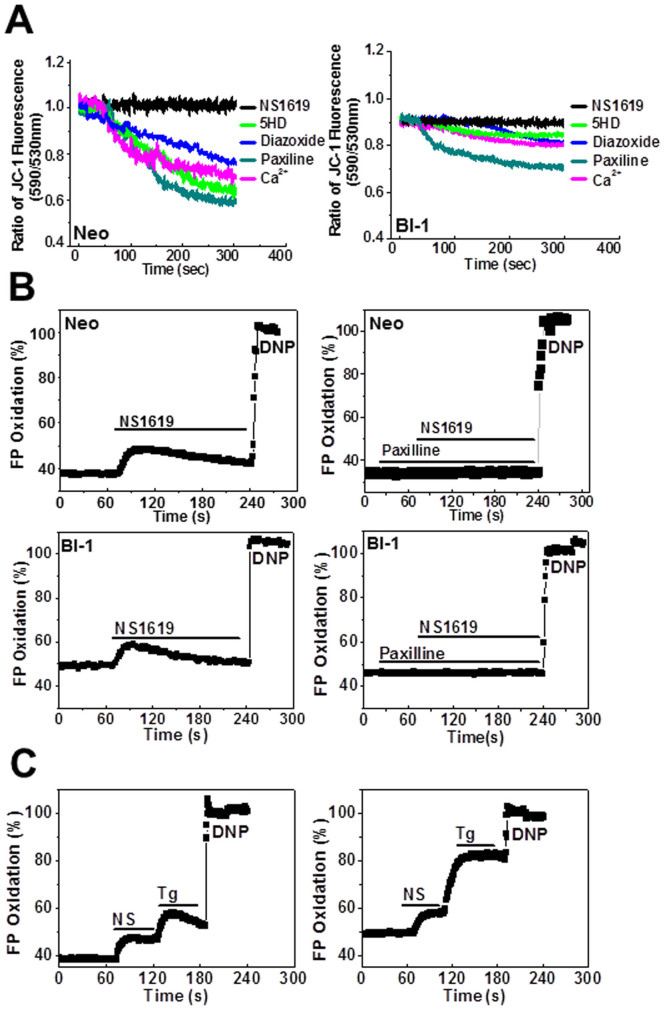
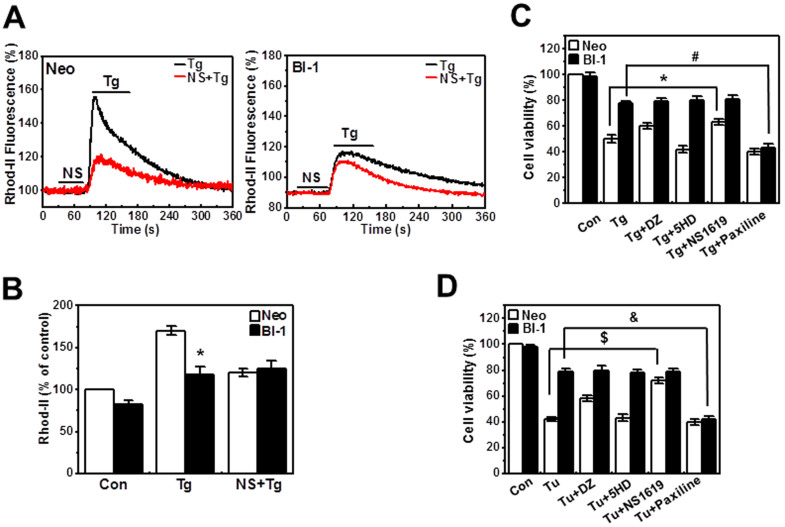
A recently studied endoplasmic reticulum (ER) stress regulator, Bax inhibitor-1 (BI-1) plays a regulatory role in mitochondrial Ca(2+) levels. In this study, we identified ER-resident and mitochondria-associated ER membrane (MAM)-resident populations of BI-1. ER stress increased mitochondrial Ca(2+) to a lesser extent in BI-1-overexpressing cells (HT1080/BI-1) than in control cells, most likely as a result of impaired mitochondrial Ca(2+) intake ability and lower basal levels of intra-ER Ca(2+). Moreover, opening of the Ca(2+)-induced mitochondrial permeability transition pore (PTP) and cytochrome c release were regulated by BI-1. In HT1080/BI-1, the basal mitochondrial membrane potential was low and also resistant to Ca(2+) compared with control cells. The activity of the mitochondrial membrane potential-dependent mitochondrial Ca(2+) intake pore, the Ca(2+) uniporter, was reduced in the presence of BI-1. This study also showed that instead of Ca(2+), other cations including K(+) enter the mitochondria of HT1080/BI-1 through mitochondrial Ca(2+)-dependent ion channels, providing a possible mechanism by which mitochondrial Ca(2+) intake is reduced, leading to cell protection. We propose a model in which BI-1-mediated sequential regulation of the mitochondrial Ca(2+) uniporter and Ca(2+)-dependent K(+) channel opening inhibits mitochondrial Ca(2+) intake, thereby inhibiting PTP function and leading to cell protection.
Figures
Similar articles
-
Bax Inhibitor-1 Is a pH-dependent regulator of Ca2+ channel activity in the endoplasmic reticulum.J Biol Chem. 2008 Jun 6;283(23):15946-55. doi: 10.1074/jbc.M800075200. Epub 2008 Mar 31. J Biol Chem. 2008. PMID: 18378668 Free PMC article.
-
Bax Inhibitor-1-mediated Ca2+ leak is decreased by cytosolic acidosis.Cell Calcium. 2013 Sep;54(3):186-92. doi: 10.1016/j.ceca.2013.06.002. Epub 2013 Jul 16. Cell Calcium. 2013. PMID: 23867001
-
Plasma membrane Ca2+-ATPase overexpression depletes both mitochondrial and endoplasmic reticulum Ca2+ stores and triggers apoptosis in insulin-secreting BRIN-BD11 cells.J Biol Chem. 2010 Oct 1;285(40):30634-43. doi: 10.1074/jbc.M110.116681. Epub 2010 Jul 26. J Biol Chem. 2010. PMID: 20660595 Free PMC article.
-
Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis.Cell Calcium. 2006 Nov-Dec;40(5-6):553-60. doi: 10.1016/j.ceca.2006.08.016. Epub 2006 Oct 30. Cell Calcium. 2006. PMID: 17074387 Free PMC article. Review.
-
Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis.Cell Calcium. 2011 Sep;50(3):222-33. doi: 10.1016/j.ceca.2011.04.007. Epub 2011 May 23. Cell Calcium. 2011. PMID: 21601280 Review.
Cited by
-
The Unfolded Protein Response: At the Intersection between Endoplasmic Reticulum Function and Mitochondrial Bioenergetics.Front Oncol. 2017 Apr 3;7:55. doi: 10.3389/fonc.2017.00055. eCollection 2017. Front Oncol. 2017. PMID: 28421160 Free PMC article. Review.
-
Transmembrane BAX inhibitor motif containing 6 suppresses presenilin-2 to preserve mitochondrial integrity after myocardial ischemia-reperfusion injury.Int J Biol Sci. 2023 Feb 13;19(4):1228-1240. doi: 10.7150/ijbs.81100. eCollection 2023. Int J Biol Sci. 2023. PMID: 36923943 Free PMC article.
-
Bax inhibitor 1 is a γ-secretase-independent presenilin-binding protein.Proc Natl Acad Sci U S A. 2019 Jan 2;116(1):141-147. doi: 10.1073/pnas.1810870116. Epub 2018 Dec 17. Proc Natl Acad Sci U S A. 2019. PMID: 30559186 Free PMC article.
-
TMBIM6/BI-1 is an intracellular environmental regulator that induces paraptosis in cancer via ROS and Calcium-activated ERAD II pathways.Oncogene. 2025 Mar;44(8):494-512. doi: 10.1038/s41388-024-03222-x. Epub 2024 Nov 29. Oncogene. 2025. PMID: 39609612 Free PMC article.
-
Mitochondria-Associated Endoplasmic Reticulum Membranes in Human Health and Diseases.MedComm (2020). 2025 Jun 27;6(7):e70259. doi: 10.1002/mco2.70259. eCollection 2025 Jul. MedComm (2020). 2025. PMID: 40584408 Free PMC article. Review.
References
-
- Berridge M. J. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–49 (2002). - PubMed
-
- Van Coppenolle F. et al. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J Cell Sci 117, 4135–42 (2004). - PubMed
-
- Ben-Hail D., Palty R. & Shoshan-Barmatz V. Measurement of Mitochondrial Ca2+ Transport Mediated by Three Transport Proteins: VDAC1, the Na+/Ca2+ Exchanger, and the Ca2+ Uniporter. Cold Spring Harb Protoc 2, 161–166 (2014). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
