

Extraribosomal l13a is a specific innate immune factor for antiviral defense
- PMID: 24899178
- PMCID: PMC4136244
- DOI: 10.1128/JVI.01129-14
Extraribosomal l13a is a specific innate immune factor for antiviral defense
Abstract
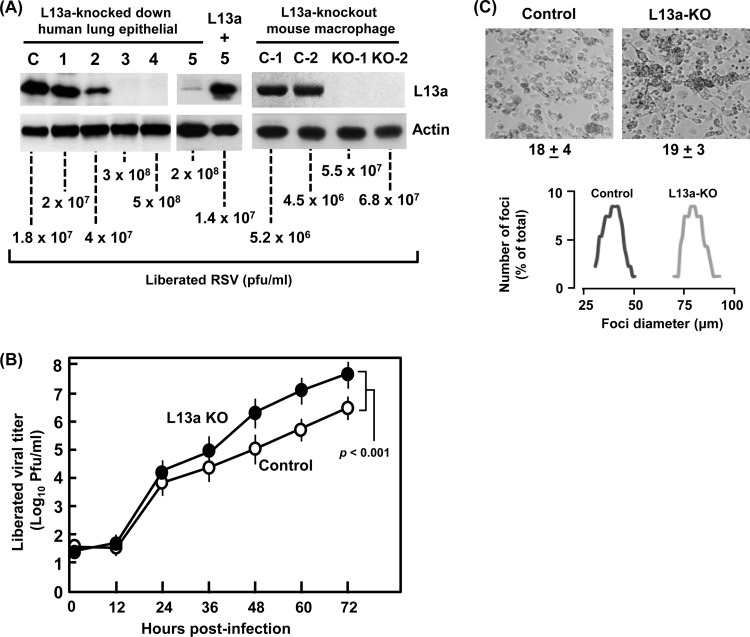
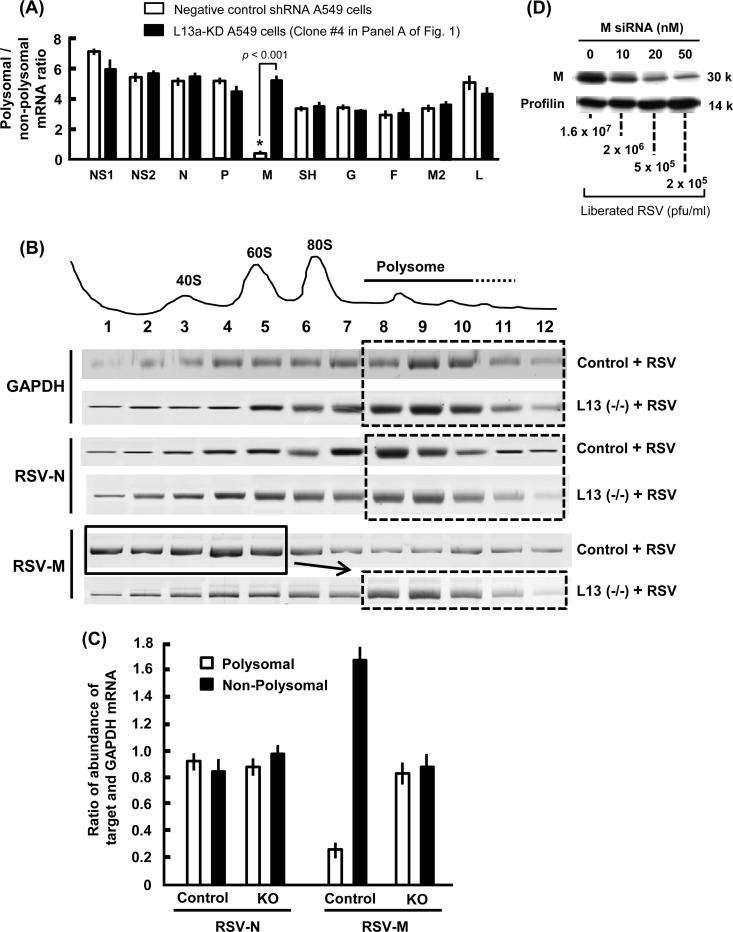
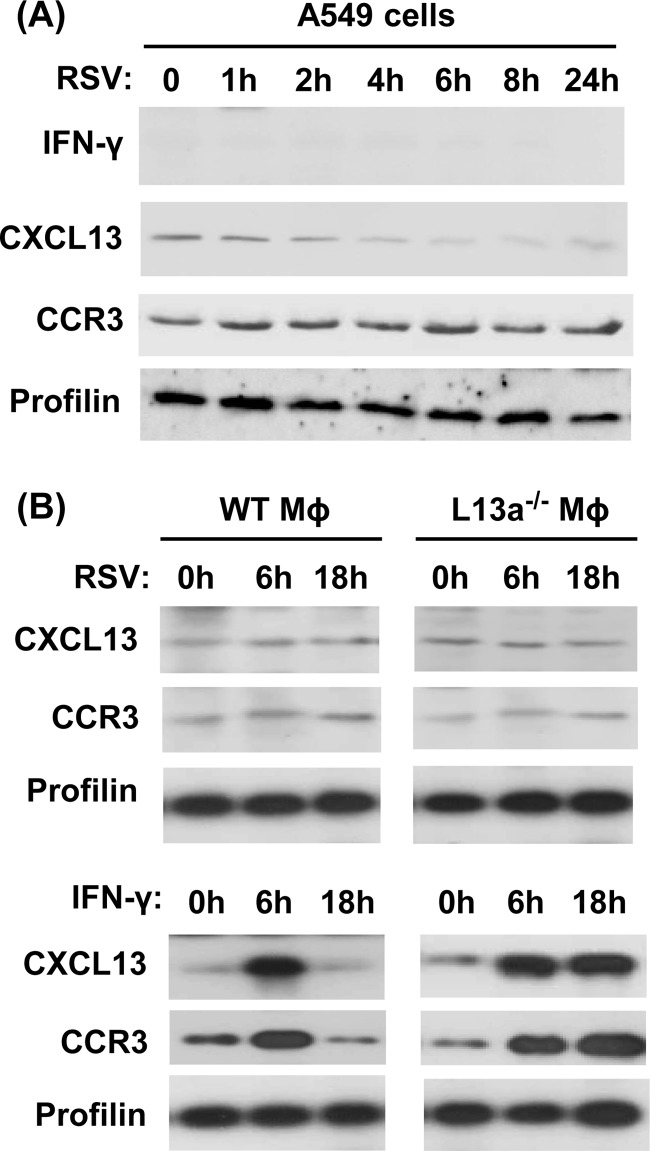
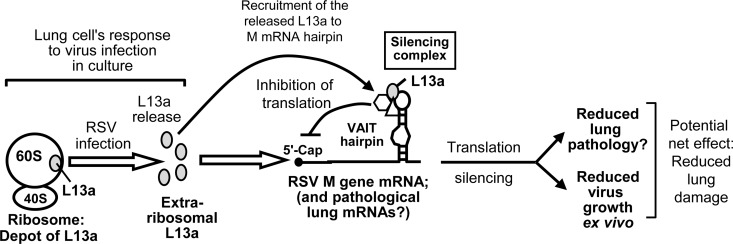
We report a novel extraribosomal innate immune function of mammalian ribosomal protein L13a, whereby it acts as an antiviral agent. We found that L13a is released from the 60S ribosomal subunit in response to infection by respiratory syncytial virus (RSV), an RNA virus of the Pneumovirus genus and a serious lung pathogen. Unexpectedly, the growth of RSV was highly enhanced in L13a-knocked-down cells of various lineages as well as in L13a knockout macrophages from mice. In all L13a-deficient cells tested, translation of RSV matrix (M) protein was specifically stimulated, as judged by a greater abundance of M protein and greater association of the M mRNA with polyribosomes, while general translation was unaffected. In silico RNA folding analysis and translational reporter assays revealed a putative hairpin in the 3'untranslated region (UTR) of M mRNA with significant structural similarity to the cellular GAIT (gamma-activated inhibitor of translation) RNA hairpin, previously shown to be responsible for assembling a large, L13a-containing ribonucleoprotein complex that promoted translational silencing in gamma interferon (IFN-γ)-activated myeloid cells. However, RNA-protein interaction studies revealed that this complex, which we named VAIT (respiratory syncytial virus-activated inhibitor of translation) is functionally different from the GAIT complex. VAIT is the first report of an extraribosomal L13a-mediated, IFN-γ-independent innate antiviral complex triggered in response to virus infection. We provide a model in which the VAIT complex strongly hinders RSV replication by inhibiting the translation of the rate-limiting viral M protein, which is a new paradigm in antiviral defense.
Importance: The innate immune mechanisms of host cells are diverse in nature and act as a broad-spectrum cellular defense against viruses. Here, we report a novel innate immune mechanism functioning against respiratory syncytial virus (RSV), in which the cellular ribosomal protein L13a is released from the large ribosomal subunit soon after infection and inhibits the translation of a specific viral mRNA, namely, that of the matrix protein M. Regarding its mechanism, we show that the recognition of a specific secondary structure in the 3' untranslated region of the M mRNA leads to translational arrest of the mRNA. We also show that the level of M protein in the infected cell is rate limiting for viral morphogenesis, providing a rationale for L13a to target the M mRNA for suppression of RSV growth. Translational silencing of a viral mRNA by a deployed ribosomal protein is a new paradigm in innate immunity.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
