

HIV-1 Vpr redirects host ubiquitination pathway
- PMID: 24899191
- PMCID: PMC4136268
- DOI: 10.1128/JVI.00619-14
HIV-1 Vpr redirects host ubiquitination pathway
Abstract
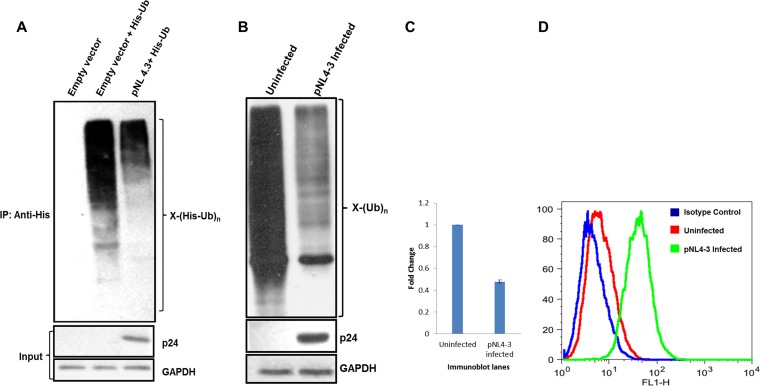
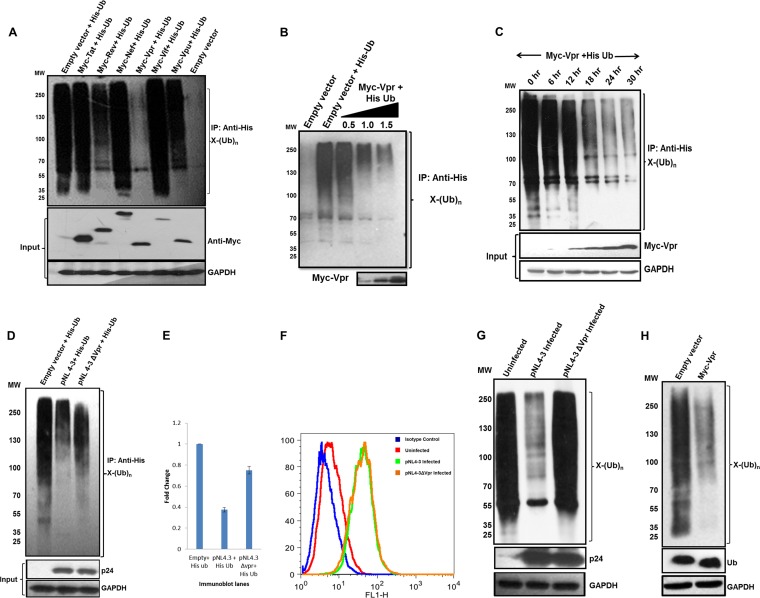
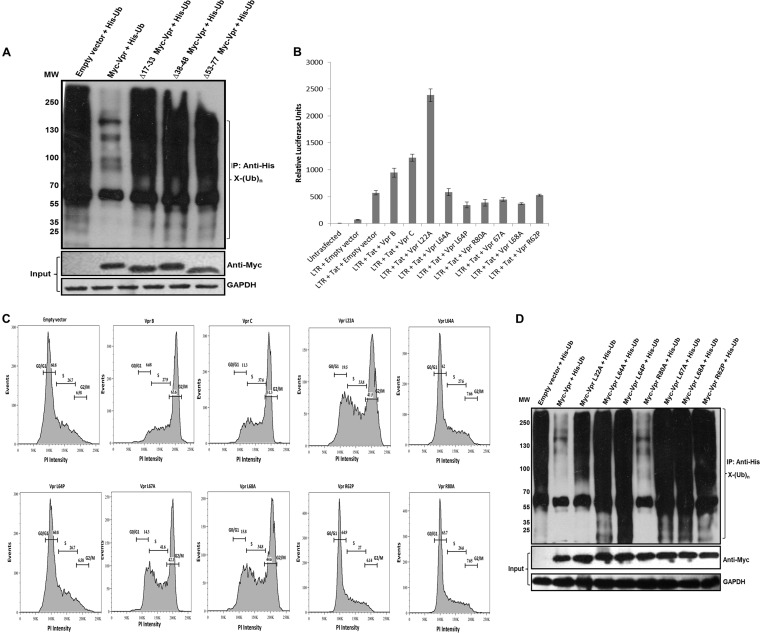
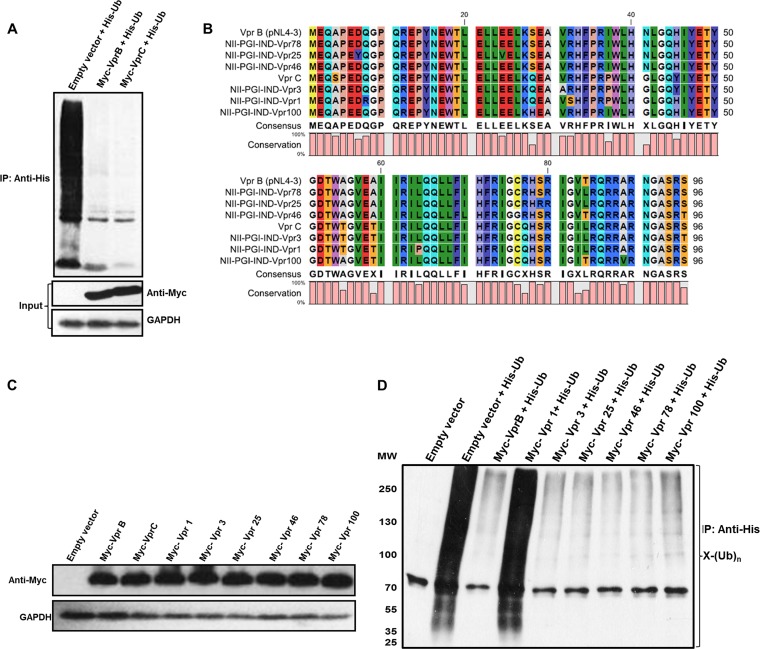
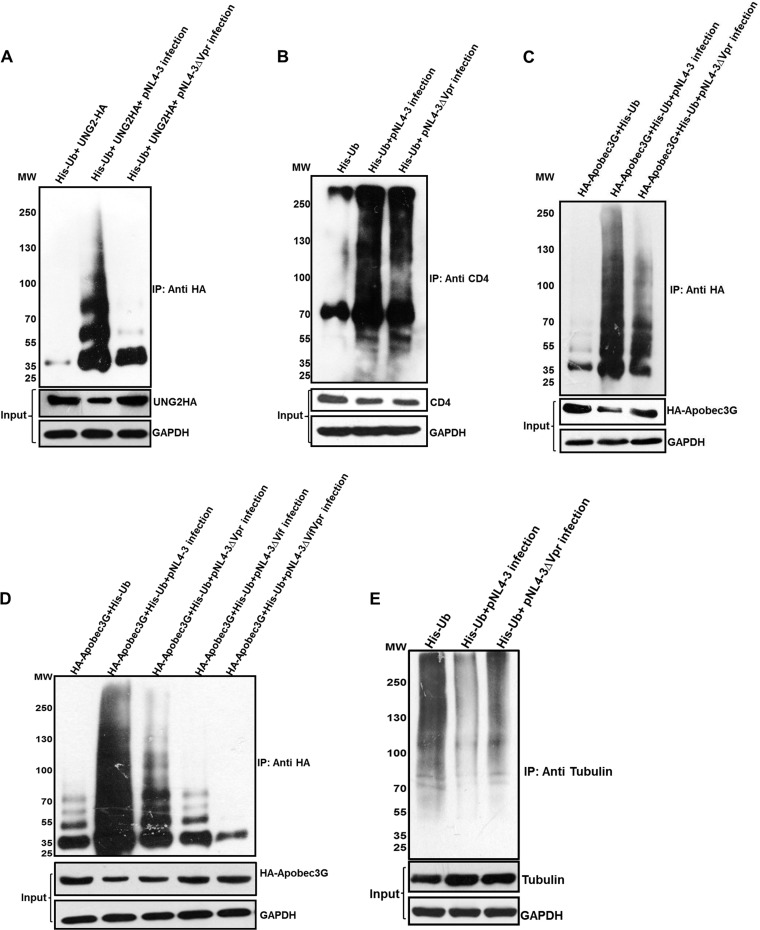
HIV-1 modulates key host cellular pathways for successful replication and pathogenesis through viral proteins. By evaluating the hijacking of the host ubiquitination pathway by HIV-1 at the whole-cell level, we now show major perturbations in the ubiquitinated pool of the host proteins post-HIV-1 infection. Our overexpression- and infection-based studies of T cells with wild-type and mutant HIV-1 proviral constructs showed that Vpr is necessary and sufficient for reducing whole-cell ubiquitination. Mutagenic analysis revealed that the three leucine-rich helical regions of Vpr are critical for this novel function of Vpr, which was independent of its other known cellular functions. We also validated that this effect of Vpr was conserved among different subtypes (subtypes B and C) and circulating recombinants from Northern India. Finally, we establish that this phenomenon is involved in HIV-1-mediated diversion of host ubiquitination machinery specifically toward the degradation of various restriction factors during viral pathogenesis.
Importance: HIV-1 is known to rely heavily on modulation of the host ubiquitin pathway, particularly for counteraction of antiretroviral restriction factors, i.e., APOBEC3G, UNG2, and BST-2, etc.; viral assembly; and release. Reports to date have focused on the molecular hijacking of the ubiquitin machinery by HIV-1 at the level of E3 ligases. Interaction of a viral protein with an E3 ligase alters its specificity to bring about selective protein ubiquitination. However, in the case of infection, multiple viral proteins can interact with this multienzyme pathway at various levels, making it much more complicated. Here, we have addressed the manipulation of ubiquitination at the whole-cell level post-HIV-1 infection. Our results show that HIV-1 Vpr is necessary and sufficient to bring about the redirection of the host ubiquitin pathway toward HIV-1-specific outcomes. We also show that the three leucine-rich helical regions of Vpr are critical for this effect and that this ability of Vpr is conserved across circulating recombinants. Our work, the first of its kind, provides novel insight into the regulation of the ubiquitin system at the whole-cell level by HIV-1.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
HIV-1 Vpr Reprograms CLR4DCAF1 E3 Ubiquitin Ligase to Antagonize Exonuclease 1-Mediated Restriction of HIV-1 Infection.mBio. 2018 Oct 23;9(5):e01732-18. doi: 10.1128/mBio.01732-18. mBio. 2018. PMID: 30352932 Free PMC article.
-
Cullin4A and cullin4B are interchangeable for HIV Vpr and Vpx action through the CRL4 ubiquitin ligase complex.J Virol. 2014 Jun;88(12):6944-58. doi: 10.1128/JVI.00241-14. Epub 2014 Apr 9. J Virol. 2014. PMID: 24719410 Free PMC article.
-
HIV-1 Vpr protein inhibits telomerase activity via the EDD-DDB1-VPRBP E3 ligase complex.J Biol Chem. 2013 May 31;288(22):15474-80. doi: 10.1074/jbc.M112.416735. Epub 2013 Apr 23. J Biol Chem. 2013. PMID: 23612978 Free PMC article.
-
CRL4-DCAF1 Ubiquitin Ligase Dependent Functions of HIV Viral Protein R and Viral Protein X.Viruses. 2024 Aug 17;16(8):1313. doi: 10.3390/v16081313. Viruses. 2024. PMID: 39205287 Free PMC article. Review.
-
The role of HIV-1 Vpr in promoting the infection of nondividing cells and in cell cycle arrest.Curr Opin HIV AIDS. 2012 Mar;7(2):187-94. doi: 10.1097/COH.0b013e32835049e0. Curr Opin HIV AIDS. 2012. PMID: 22274659 Free PMC article. Review.
Cited by
-
Proteasomal Degradation Machinery: Favorite Target of HIV-1 Proteins.Front Microbiol. 2018 Nov 21;9:2738. doi: 10.3389/fmicb.2018.02738. eCollection 2018. Front Microbiol. 2018. PMID: 30524389 Free PMC article. Review.
-
Network of MicroRNAs Mediate Translational Repression of Bone Morphogenetic Protein Receptor-2: Involvement in HIV-Associated Pulmonary Vascular Remodeling.J Am Heart Assoc. 2018 Feb 25;7(5):e008472. doi: 10.1161/JAHA.117.008472. J Am Heart Assoc. 2018. PMID: 29478969 Free PMC article.
-
Impact of HIV-1 Vpr manipulation of the DNA repair enzyme UNG2 on B lymphocyte class switch recombination.J Transl Med. 2020 Aug 10;18(1):310. doi: 10.1186/s12967-020-02478-7. J Transl Med. 2020. PMID: 32778120 Free PMC article.
-
HIV-1 Vpr Functions in Primary CD4+ T Cells.Viruses. 2024 Mar 9;16(3):420. doi: 10.3390/v16030420. Viruses. 2024. PMID: 38543785 Free PMC article. Review.
-
Ubiquitination and SUMOylation in HIV Infection: Friends and Foes.Curr Issues Mol Biol. 2020;35:159-194. doi: 10.21775/cimb.035.159. Epub 2019 Aug 18. Curr Issues Mol Biol. 2020. PMID: 31422939 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
