

A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples
- PMID: 24899342
- PMCID: PMC4079973
- DOI: 10.1101/gr.171934.113
A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples
Abstract
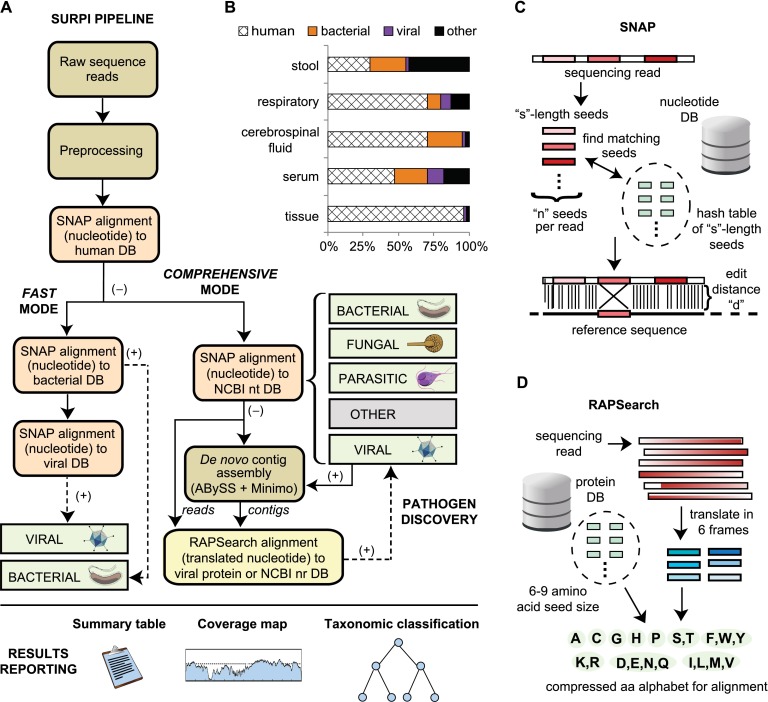
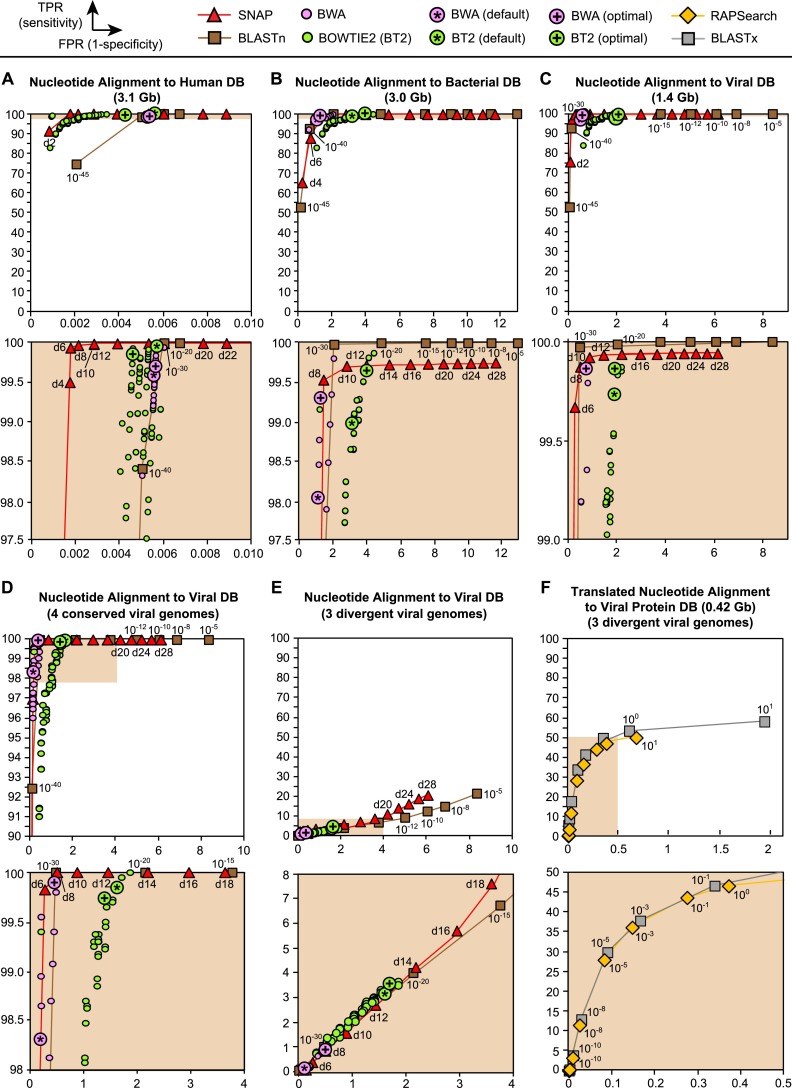
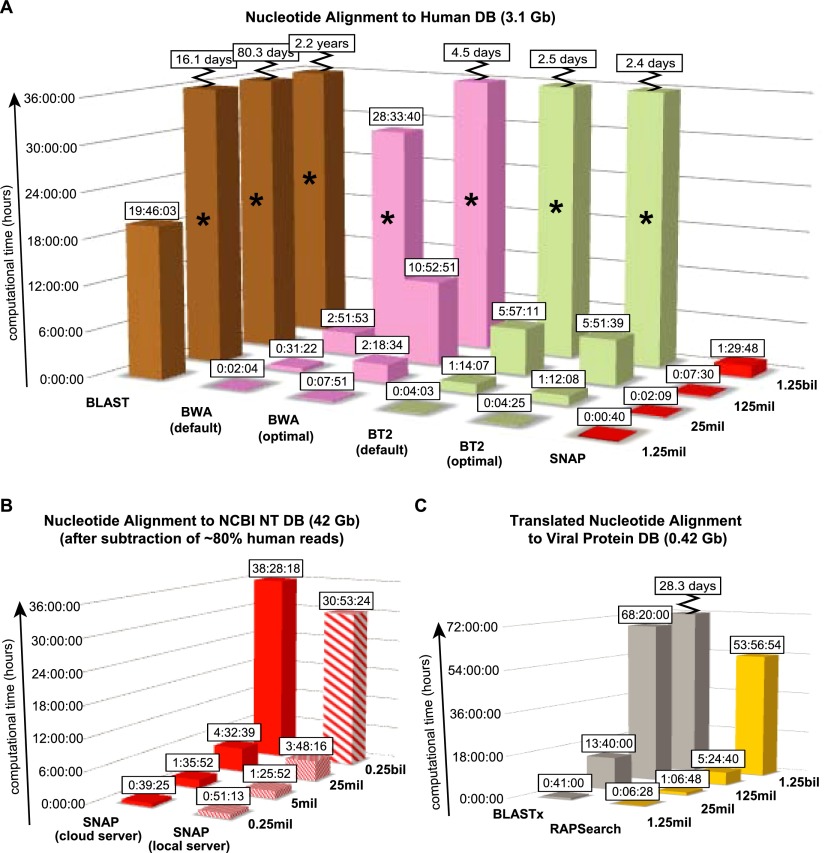
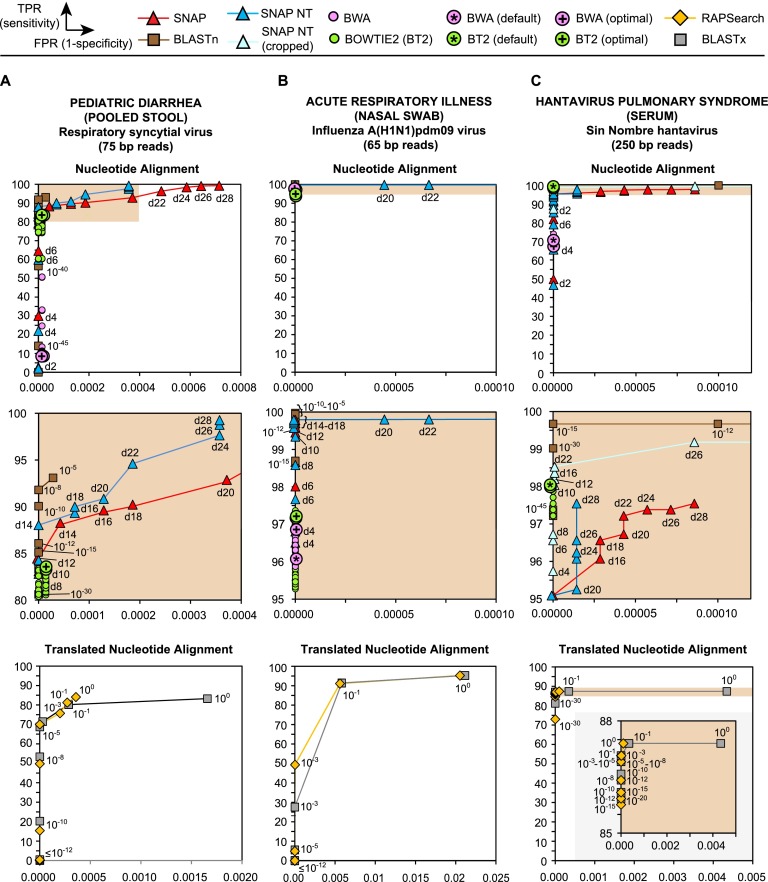
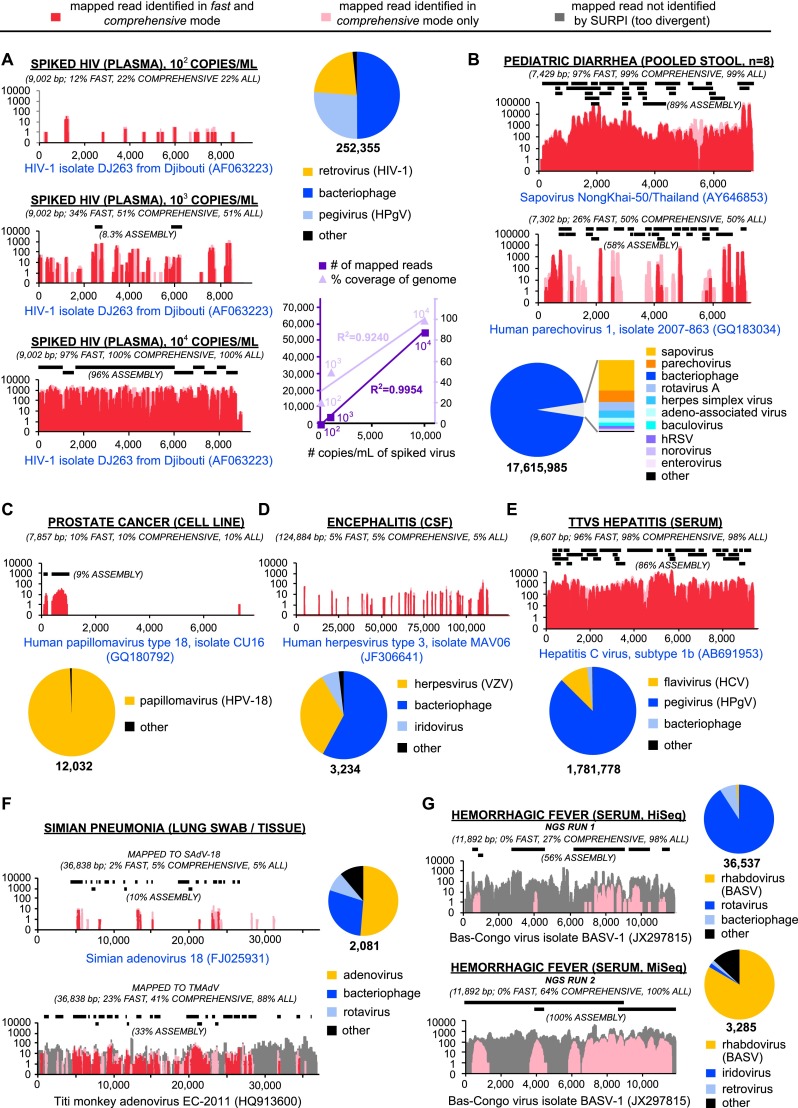
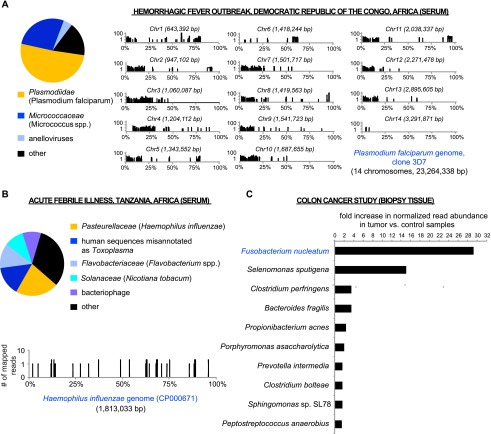
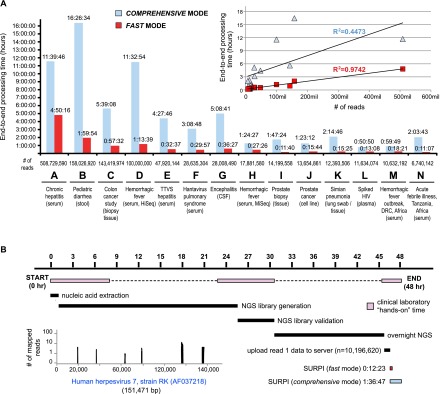
Unbiased next-generation sequencing (NGS) approaches enable comprehensive pathogen detection in the clinical microbiology laboratory and have numerous applications for public health surveillance, outbreak investigation, and the diagnosis of infectious diseases. However, practical deployment of the technology is hindered by the bioinformatics challenge of analyzing results accurately and in a clinically relevant timeframe. Here we describe SURPI ("sequence-based ultrarapid pathogen identification"), a computational pipeline for pathogen identification from complex metagenomic NGS data generated from clinical samples, and demonstrate use of the pipeline in the analysis of 237 clinical samples comprising more than 1.1 billion sequences. Deployable on both cloud-based and standalone servers, SURPI leverages two state-of-the-art aligners for accelerated analyses, SNAP and RAPSearch, which are as accurate as existing bioinformatics tools but orders of magnitude faster in performance. In fast mode, SURPI detects viruses and bacteria by scanning data sets of 7-500 million reads in 11 min to 5 h, while in comprehensive mode, all known microorganisms are identified, followed by de novo assembly and protein homology searches for divergent viruses in 50 min to 16 h. SURPI has also directly contributed to real-time microbial diagnosis in acutely ill patients, underscoring its potential key role in the development of unbiased NGS-based clinical assays in infectious diseases that demand rapid turnaround times.
© 2014 Naccache et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Akobeng AK 2007. Understanding diagnostic tests 3: receiver operating characteristic curves. Acta Paediatr 96: 644–647 - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410 - PubMed
-
- Bloch KC, Glaser C 2007. Diagnostic approaches for patients with suspected encephalitis. Curr Infect Dis Rep 9: 315–322 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical