

Troponin I-interacting protein kinase: a novel cardiac-specific kinase, emerging as a molecular target for the treatment of cardiac disease
- PMID: 24899531
- PMCID: PMC4151348
- DOI: 10.1253/circj.cj-14-0543
Troponin I-interacting protein kinase: a novel cardiac-specific kinase, emerging as a molecular target for the treatment of cardiac disease
Abstract
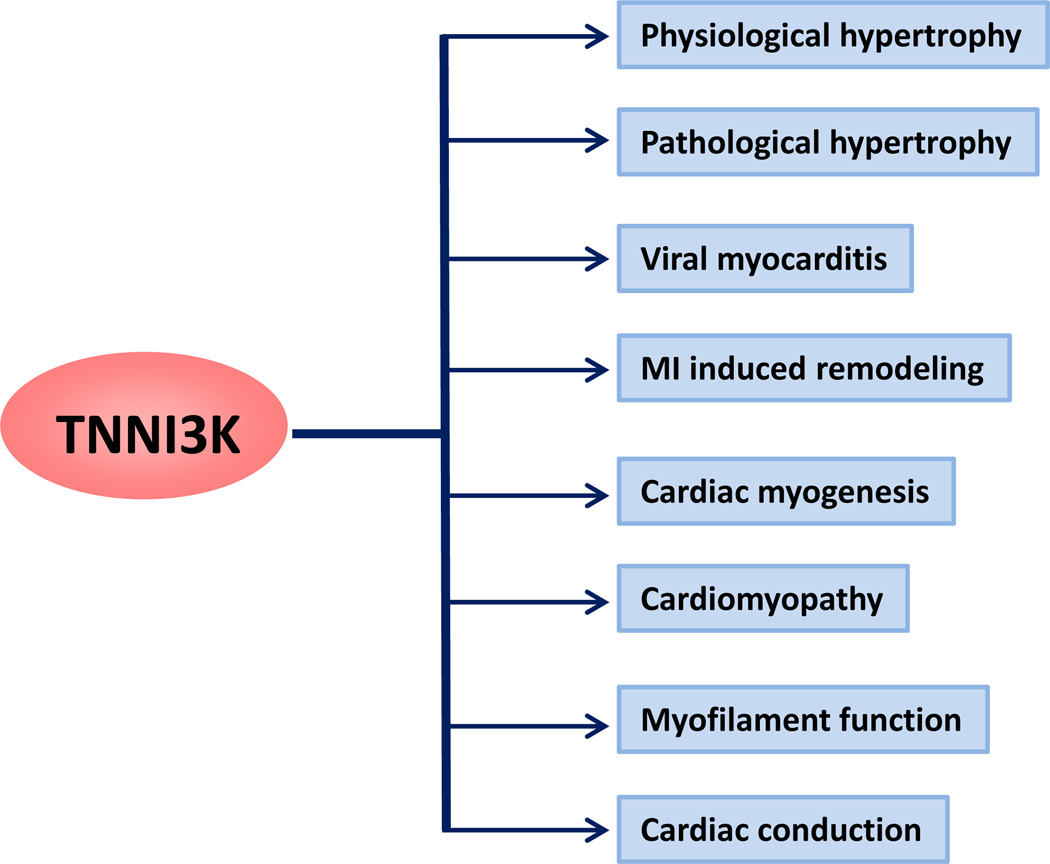
Coronary artery disease is the leading cause of death and disability worldwide. In patients with acute coronary syndromes, timely and effective myocardial reperfusion by percutaneous coronary intervention is the primary treatment of choice to minimize the ischemic injury and limit the size of the myocardial infarction (MI). However, reperfusion can itself promote cardiomyocyte death, which leads to cardiac dysfunction via reperfusion injury. The molecular mechanisms of ischemia-reperfusion (IR) injury are not completely understood and new drug targets are needed. Recently, we reported that cardiac troponin I-interacting protein kinase (TNNI3K), a cardiomyocyte-specific kinase, promotes IR injury via profound oxidative stress, thereby promoting cardiomyocyte death. By using novel genetic animal models and newly developed small-molecule TNNI3K inhibitors, we demonstrated that TNNI3K-mediated IR injury occurs through impaired mitochondrial function and is in part dependent on p38 MAPK. Here we discuss the emerging role of TNNI3K as a promising new drug target to limit IR-induced myocardial injury. We will also examine the underlying mechanisms that drive the profoundly reduced infarct size in mice in whichTNNI3Kis specifically deleted in cardiomyocytes. Because TNNI3K is a cardiac-specific kinase, it could be an ideal molecular target, as inhibiting it would have little or no effect on other organ systems, a serious problem associated with the use of kinase inhibitors targeting kinases that are more widely expressed.
Figures


Similar articles
-
Inhibition of the cardiomyocyte-specific kinase TNNI3K limits oxidative stress, injury, and adverse remodeling in the ischemic heart.Sci Transl Med. 2013 Oct 16;5(207):207ra141. doi: 10.1126/scitranslmed.3006479. Sci Transl Med. 2013. PMID: 24132636 Free PMC article.
-
DNA-PKcs promotes cardiac ischemia reperfusion injury through mitigating BI-1-governed mitochondrial homeostasis.Basic Res Cardiol. 2020 Jan 9;115(2):11. doi: 10.1007/s00395-019-0773-7. Basic Res Cardiol. 2020. PMID: 31919590
-
Overexpression of TNNI3K, a cardiac-specific MAP kinase, promotes P19CL6-derived cardiac myogenesis and prevents myocardial infarction-induced injury.Am J Physiol Heart Circ Physiol. 2008 Aug;295(2):H708-16. doi: 10.1152/ajpheart.00252.2008. Epub 2008 Jun 13. Am J Physiol Heart Circ Physiol. 2008. PMID: 18552163
-
Mitochondria as a therapeutic target for cardiac ischemia‑reperfusion injury (Review).Int J Mol Med. 2021 Feb;47(2):485-499. doi: 10.3892/ijmm.2020.4823. Epub 2020 Dec 16. Int J Mol Med. 2021. PMID: 33416090 Free PMC article. Review.
-
The roles of PKC-δ and PKC-ε in myocardial ischemia/reperfusion injury.Pharmacol Res. 2021 Aug;170:105716. doi: 10.1016/j.phrs.2021.105716. Epub 2021 Jun 5. Pharmacol Res. 2021. PMID: 34102229 Review.
Cited by
-
A Novel Missense Mutation in TNNI3K Causes Recessively Inherited Cardiac Conduction Disease in a Consanguineous Pakistani Family.Genes (Basel). 2021 Aug 21;12(8):1282. doi: 10.3390/genes12081282. Genes (Basel). 2021. PMID: 34440456 Free PMC article.
-
Mycn ameliorates cardiac hypertrophy-induced heart failure in mice by mediating the USP2/JUP/Akt/β-catenin cascade.BMC Cardiovasc Disord. 2024 Jan 31;24(1):82. doi: 10.1186/s12872-024-03748-8. BMC Cardiovasc Disord. 2024. PMID: 38297207 Free PMC article.
-
Genetic analyses of the electrocardiographic QT interval and its components identify additional loci and pathways.Nat Commun. 2022 Sep 1;13(1):5144. doi: 10.1038/s41467-022-32821-z. Nat Commun. 2022. PMID: 36050321 Free PMC article.
-
Silencing Cardiac Troponin I-Interacting Kinase Reduces Lipopolysaccharide-Induced Sepsis-Induced Myocardial Dysfunction in Rat by Regulating Apoptosis-Related Proteins.Biomed Res Int. 2021 May 27;2021:5520051. doi: 10.1155/2021/5520051. eCollection 2021. Biomed Res Int. 2021. PMID: 34136567 Free PMC article.
-
Refined Phylogenetic Ortholog Inference Reveals Coevolutionary Expansion of the MAPK Signaling Network Through Finetuning of Pathway Specificity.J Mol Evol. 2025 Jun;93(3):423-440. doi: 10.1007/s00239-025-10254-8. Epub 2025 May 30. J Mol Evol. 2025. PMID: 40447942
References
-
- Gerczuk PZ, Kloner RA. An update on cardioprotection: A review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59:969–978. - PubMed
-
- Kloner RA. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ Res. 2013;113:451–463. - PubMed
-
- Minamino T. Cardioprotection from ischemia/reperfusion injury: Basic and translational research. Circ J. 2012;76:1074–1082. - PubMed
-
- Frohlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ. Myocardial reperfusion injury: Looking beyond primary pci. Eur Heart J. 2013;34:1714–1722. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials