

Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart
- PMID: 24899689
- PMCID: PMC4153405
- DOI: 10.1161/CIRCULATIONAHA.113.008364
Cardiac fibroblast glycogen synthase kinase-3β regulates ventricular remodeling and dysfunction in ischemic heart
Abstract
Background: Myocardial infarction-induced remodeling includes chamber dilatation, contractile dysfunction, and fibrosis. Of these, fibrosis is the least understood. After myocardial infarction, activated cardiac fibroblasts deposit extracellular matrix. Current therapies to prevent fibrosis are inadequate, and new molecular targets are needed.
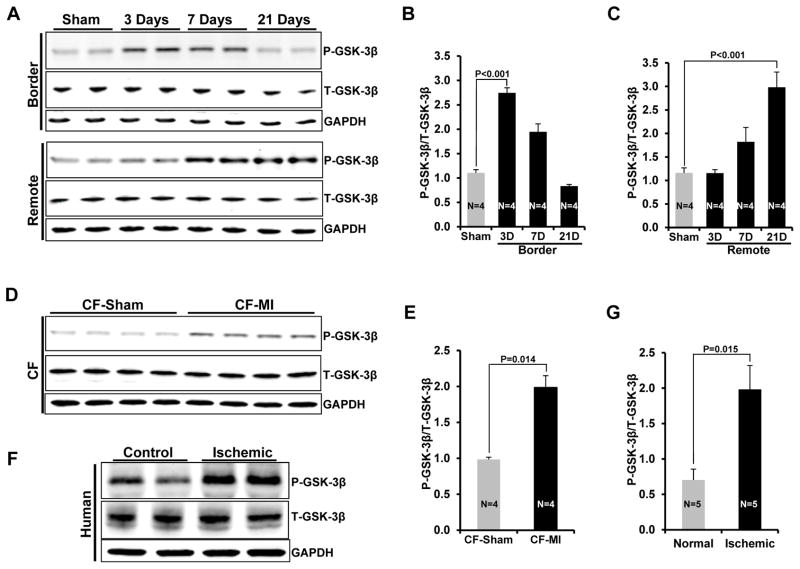
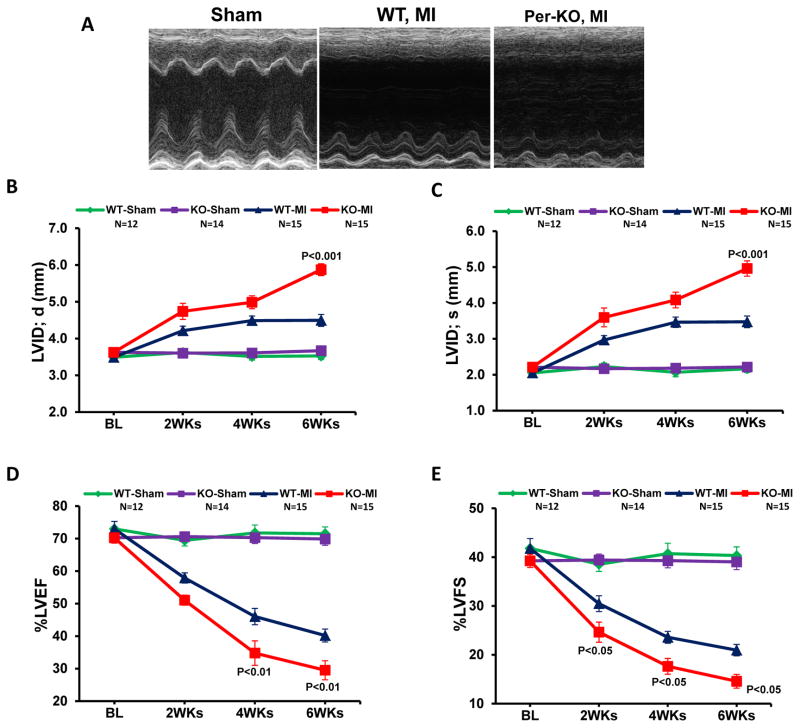
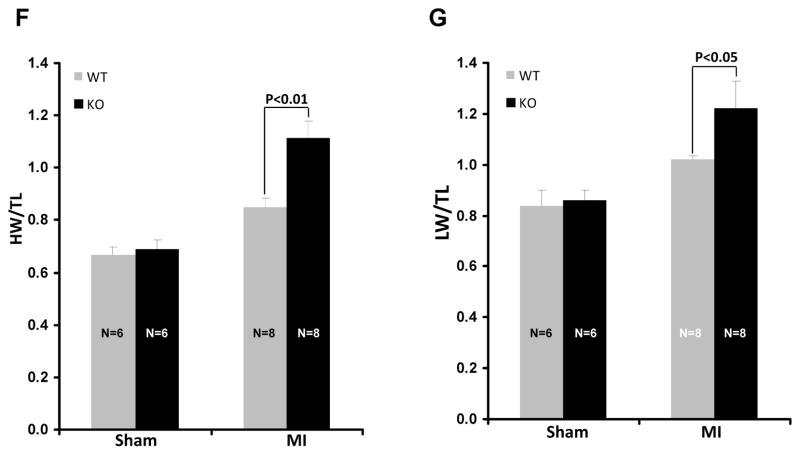
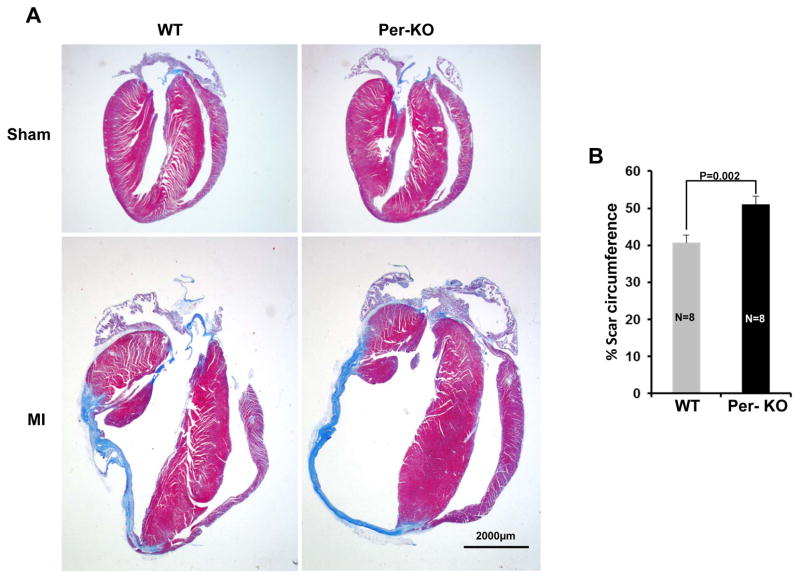
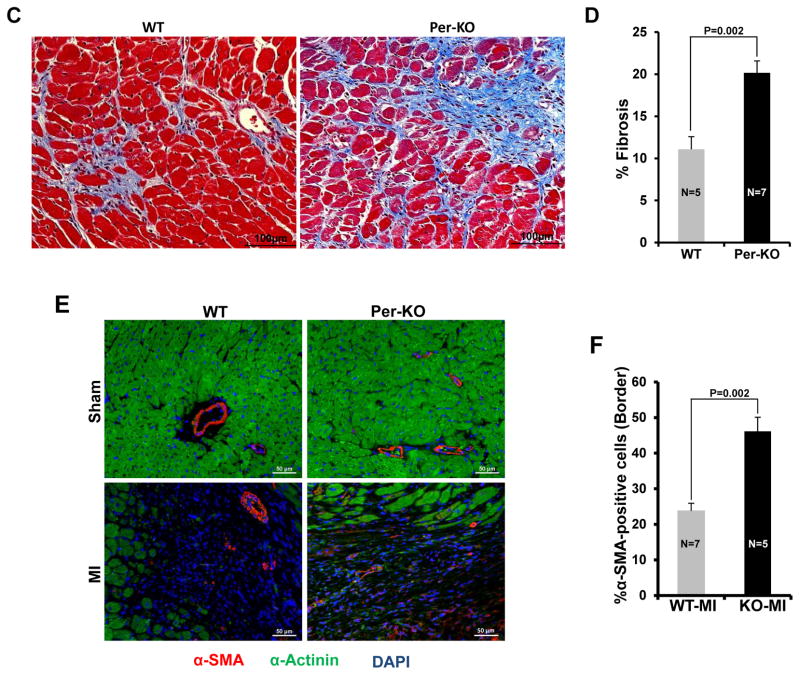
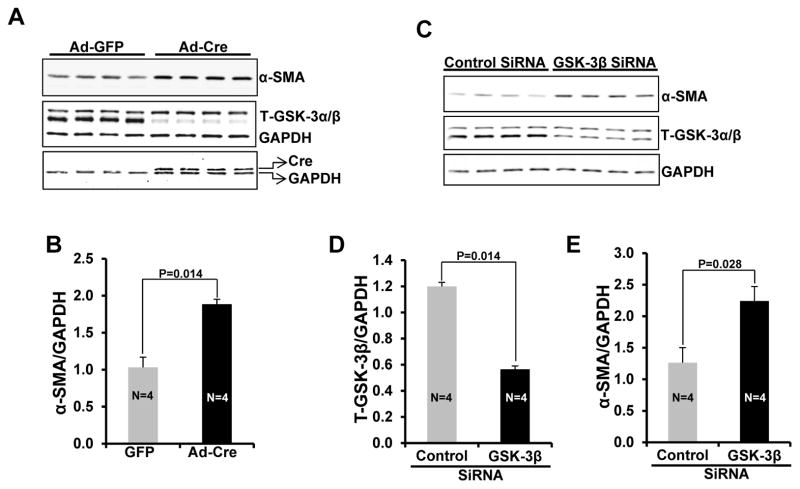
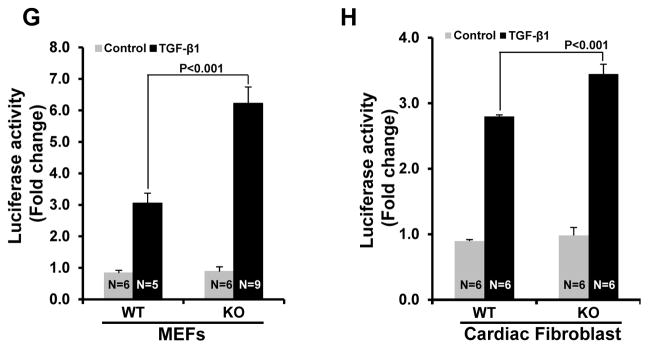
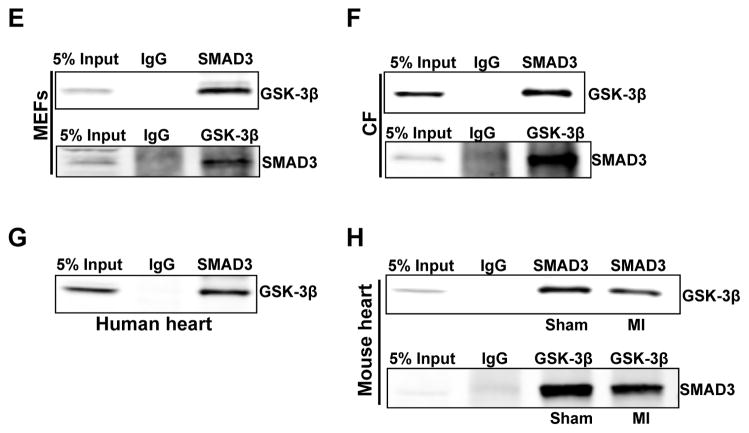
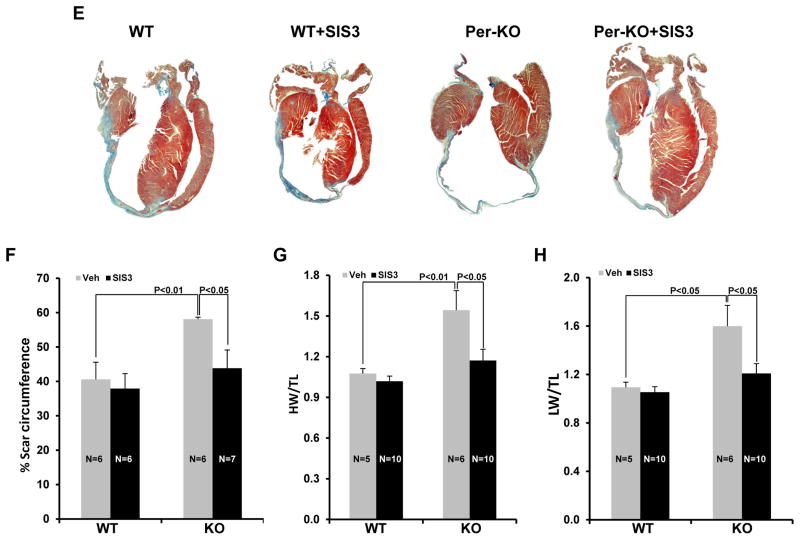
Methods and results: Herein we report that glycogen synthase kinase-3β (GSK-3β) is phosphorylated (inhibited) in fibrotic tissues from ischemic human and mouse heart. Using 2 fibroblast-specific GSK-3β knockout mouse models, we show that deletion of GSK-3β in cardiac fibroblasts leads to fibrogenesis, left ventricular dysfunction, and excessive scarring in the ischemic heart. Deletion of GSK-3β induces a profibrotic myofibroblast phenotype in isolated cardiac fibroblasts, in post-myocardial infarction hearts, and in mouse embryonic fibroblasts deleted for GSK-3β. Mechanistically, GSK-3β inhibits profibrotic transforming growth factor-β1/SMAD-3 signaling via interactions with SMAD-3. Moreover, deletion of GSK-3β resulted in the significant increase of SMAD-3 transcriptional activity. This pathway is central to the pathology because a small-molecule inhibitor of SMAD-3 largely prevented fibrosis and limited left ventricular remodeling.
Conclusions: These studies support targeting GSK-3β in myocardial fibrotic disorders and establish critical roles of cardiac fibroblasts in remodeling and ventricular dysfunction.
Keywords: fibroblasts; fibrosis; glycogen synthase kinase 3 beta; hypertrophy; myocardial infarction.
© 2014 American Heart Association, Inc.
Figures






References
-
- Lijnen PJ, Maharani T, Finahari N, Prihadi JS. Serum collagen markers and heart failure. Cardiovasc Hematol Disord Drug Targets. 2012;12:51–55. - PubMed
-
- Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15–26. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
