

Identification of divergent protein domains by combining HMM-HMM comparisons and co-occurrence detection
- PMID: 24901648
- PMCID: PMC4046975
- DOI: 10.1371/journal.pone.0095275
Identification of divergent protein domains by combining HMM-HMM comparisons and co-occurrence detection
Abstract
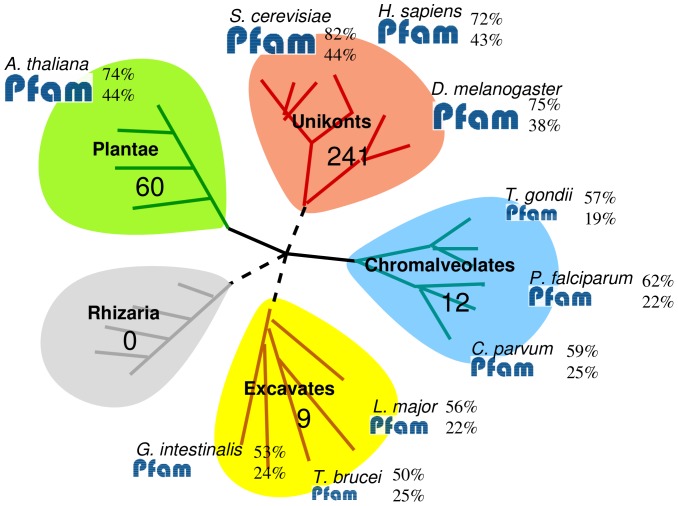
Identification of protein domains is a key step for understanding protein function. Hidden Markov Models (HMMs) have proved to be a powerful tool for this task. The Pfam database notably provides a large collection of HMMs which are widely used for the annotation of proteins in sequenced organisms. This is done via sequence/HMM comparisons. However, this approach may lack sensitivity when searching for domains in divergent species. Recently, methods for HMM/HMM comparisons have been proposed and proved to be more sensitive than sequence/HMM approaches in certain cases. However, these approaches are usually not used for protein domain discovery at a genome scale, and the benefit that could be expected from their utilization for this problem has not been investigated. Using proteins of P. falciparum and L. major as examples, we investigate the extent to which HMM/HMM comparisons can identify new domain occurrences not already identified by sequence/HMM approaches. We show that although HMM/HMM comparisons are much more sensitive than sequence/HMM comparisons, they are not sufficiently accurate to be used as a standalone complement of sequence/HMM approaches at the genome scale. Hence, we propose to use domain co-occurrence--the general domain tendency to preferentially appear along with some favorite domains in the proteins--to improve the accuracy of the approach. We show that the combination of HMM/HMM comparisons and co-occurrence domain detection boosts protein annotations. At an estimated False Discovery Rate of 5%, it revealed 901 and 1098 new domains in Plasmodium and Leishmania proteins, respectively. Manual inspection of part of these predictions shows that it contains several domain families that were missing in the two organisms. All new domain occurrences have been integrated in the EuPathDomains database, along with the GO annotations that can be deduced.
Conflict of interest statement
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
