

Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis
- PMID: 24902902
- PMCID: PMC4211369
- DOI: 10.1038/cdd.2014.77
Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis
Abstract
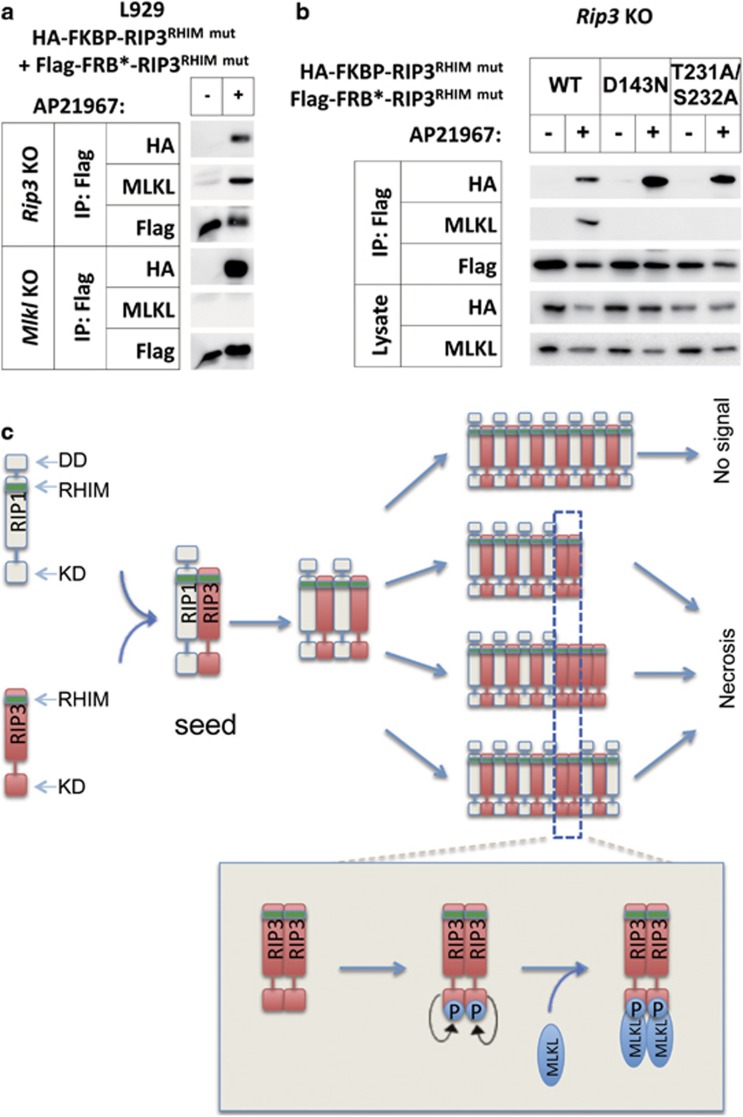
Necroptosis is mediated by a signaling complex called necrosome, containing receptor-interacting protein (RIP)1, RIP3, and mixed-lineage kinase domain-like (MLKL). It is known that RIP1 and RIP3 form heterodimeric filamentous scaffold in necrosomes through their RIP homotypic interaction motif (RHIM) domain-mediated oligomerization, but the signaling events based on this scaffold has not been fully addressed. By using inducible dimer systems we found that RIP1-RIP1 interaction is dispensable for necroptosis; RIP1-RIP3 interaction is required for necroptosis signaling, but there is no necroptosis if no additional RIP3 protein is recruited to the RIP1-RIP3 heterodimer, and the interaction with RIP1 promotes the RIP3 to recruit other RIP3; RIP3-RIP3 interaction is required for necroptosis and RIP3-RIP3 dimerization is sufficient to induce necroptosis; and RIP3 dimer-induced necroptosis requires MLKL. We further show that RIP3 oligomer is not more potent than RIP3 dimer in triggering necroptosis, suggesting that RIP3 homo-interaction in the complex, rather than whether RIP3 has formed homo polymer, is important for necroptosis. RIP3 dimerization leads to RIP3 intramolecule autophosphorylation, which is required for the recruitment of MLKL. Interestingly, phosphorylation of one of RIP3 in the dimer is sufficient to induce necroptosis. As RIP1-RIP3 heterodimer itself cannot induce necroptosis, the RIP1-RIP3 heterodimeric amyloid fibril is unlikely to directly propagate necroptosis. We propose that the signaling events after the RIP1-RIP3 amyloid complex assembly are the recruitment of free RIP3 by the RIP3 in the amyloid scaffold followed by autophosphorylation of RIP3 and subsequent recruitment of MLKL by RIP3 to execute necroptosis.
Figures





References
-
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. - PubMed
-
- Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. - PubMed
-
- Han J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12:1143–1149. - PubMed
-
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. - PubMed
-
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
