

It's MORe exciting than mu: crosstalk between mu opioid receptors and glutamatergic transmission in the mesolimbic dopamine system
- PMID: 24904419
- PMCID: PMC4034717
- DOI: 10.3389/fphar.2014.00116
It's MORe exciting than mu: crosstalk between mu opioid receptors and glutamatergic transmission in the mesolimbic dopamine system
Abstract
Opioids selective for the G protein-coupled mu opioid receptor (MOR) produce potent analgesia and euphoria. Heroin, a synthetic opioid, is considered one of the most addictive substances, and the recent exponential rise in opioid addiction and overdose deaths has made treatment development a national public health priority. Existing medications (methadone, buprenorphine, and naltrexone), when combined with psychosocial therapies, have proven efficacy in reducing aspects of opioid addiction. Unfortunately, these medications have critical limitations including those associated with opioid agonist therapies (e.g., sustained physiological dependence and opioid withdrawal leading to high relapse rates upon discontinuation), non-adherence to daily dosing, and non-renewal of monthly injection with extended-release naltrexone. Furthermore, current medications fail to ameliorate key aspects of addiction such as powerful conditioned associations that trigger relapse (e.g., cues, stress, the drug itself). Thus, there is a need for developing novel treatments that target neural processes corrupted with chronic opioid use. This requires a basic understanding of molecular and cellular mechanisms underlying effects of opioids on synaptic transmission and plasticity within reward-related neural circuits. The focus of this review is to discuss how crosstalk between MOR-associated G protein signaling and glutamatergic neurotransmission leads to immediate and long-term effects on emotional states (e.g., euphoria, depression) and motivated behavior (e.g., drug-seeking, relapse). Our goal is to integrate findings on how opioids modulate synaptic release of glutamate and postsynaptic transmission via α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and N-methyl-D-aspartate receptors in the nucleus accumbens and ventral tegmental area with the clinical (neurobehavioral) progression of opioid dependence, as well as to identify gaps in knowledge that can be addressed in future studies.
Keywords: AMPA; GluR1; NMDA; heroin; morphine; opioid withdrawal syndrome; plasticity.
Figures
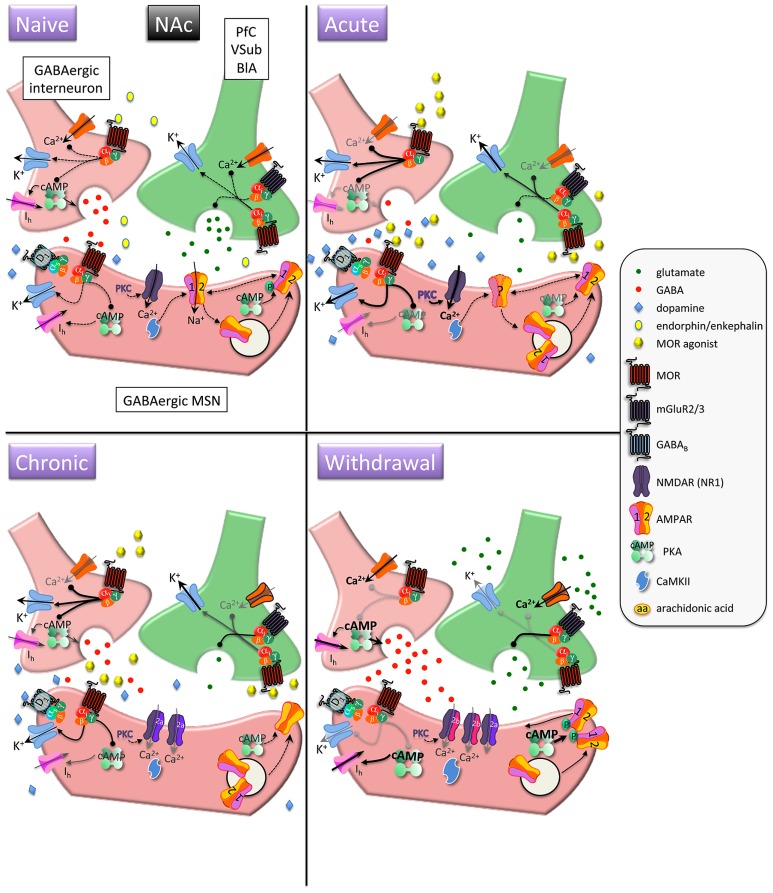
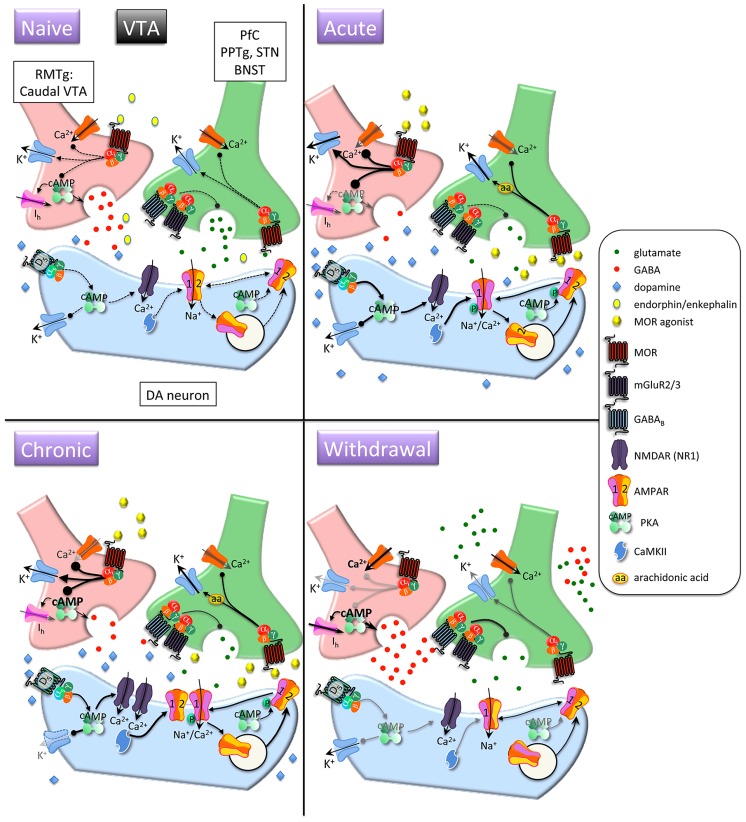
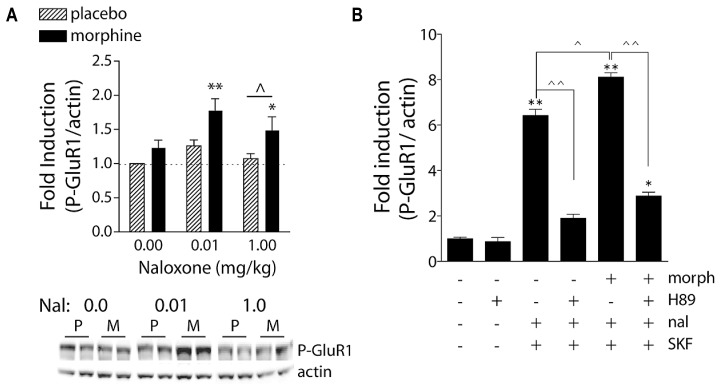
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials