

Review
doi: 10.3389/fgene.2014.00157.
eCollection 2014.
Quality control on the frontier
Affiliations
- PMID: 24904650
- PMCID: PMC4033843
- DOI: 10.3389/fgene.2014.00157
Item in Clipboard
Review
Quality control on the frontier
Front Genet.
.
Abstract
In the world of high-throughput sequencing there are numerous challenges to effective data quality control. There are no single quality metrics which are appropriate in all conditions. Here we detail the different open source software used at the Exeter Sequencing Service to provide generic quality control information, as well as more specific metrics for genomic and transcriptomic libraries run on Illumina platforms.
Keywords: Illumina; best practice; core-facility; quality control; sequencing.
Figures
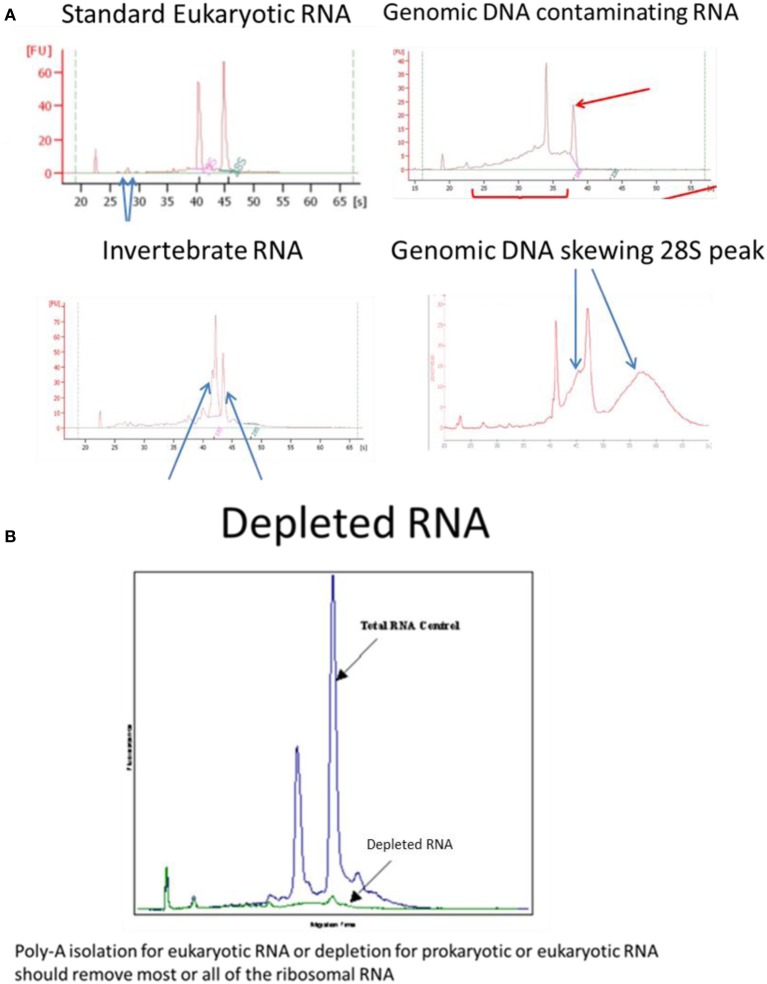
Examples of Bioanalyser assay interpretation for a variety of RNAs. (A) Standard Eukaryotic RNA shows a 28S rRNA band at 4.5 kb that should be twice the intensity of the 18S rRNA band at 1.9 kb (human) resulting in a RIN = 8.0–10.0. Small peaks are sometimes present after the marker that represent 5S and 5.8S subunits, tRNAs and small RNA fragments about 100 bp; these are more obvious when using phenol or trizol exterection methods, QIagen columns will generally remove small RNAs. When degraded 28S RNA is reduced and more fragments are detected around the 18S RNA subunit resulting in RIN = 6.4, which is below the quality required for high throughput DNA sequencing. Invertebrate RNA results in fragmentation of the 28S rRNA into two bands that co-migrate with the 18S rRNA resulting in aberrant RIN score of <8.0 although the mRNA is unaffected and suitable for sequencing. Genomic DNA can skew the 28S RNA peak but can easily be remedied by RNAse-free DNase1 digestion. (B) Ribosomal RNA removal by isolation of poly-A-RNA assessed by Bioanalyser RNA assay.
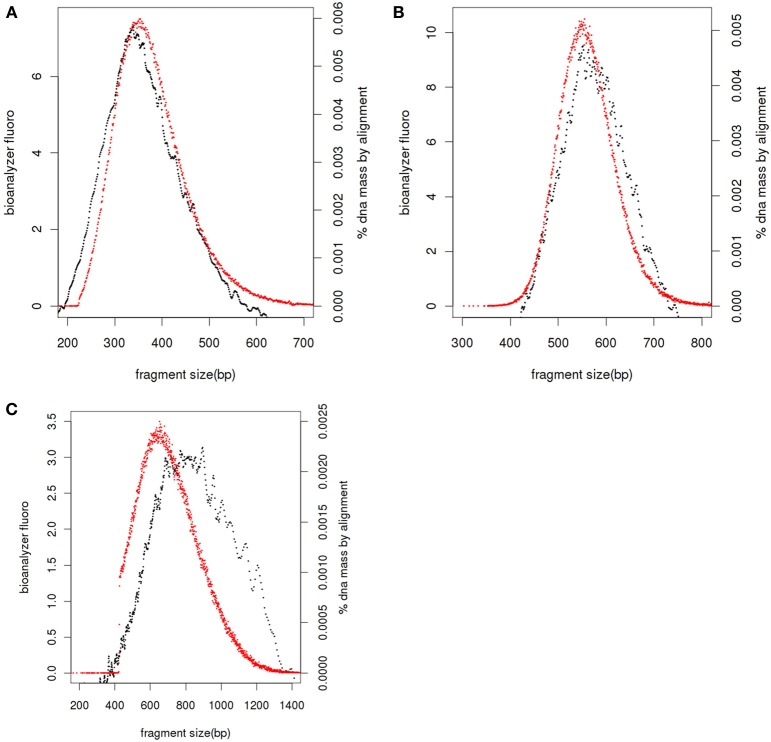
Library fragment size distribution. Bioanalyser fluorescence values (black), realignment of paired end reads against reference genome or de novo assembly and adjusted by 126 bases to account for adapters (red) for libraries with average sizes of 360 bases (A), 550 bases (B), and 810 bases (C).
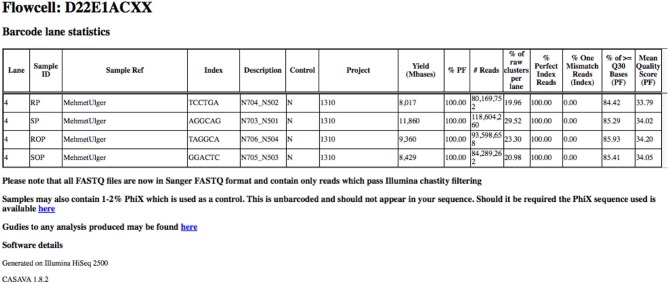
Basic read metrics extracted on a per-project basis. Basic read metrics extracted on a per-sample basis from the Illumina Demultiplex_stats.html file produced by the bcl2fastq pipeline. Additional information has also been added.
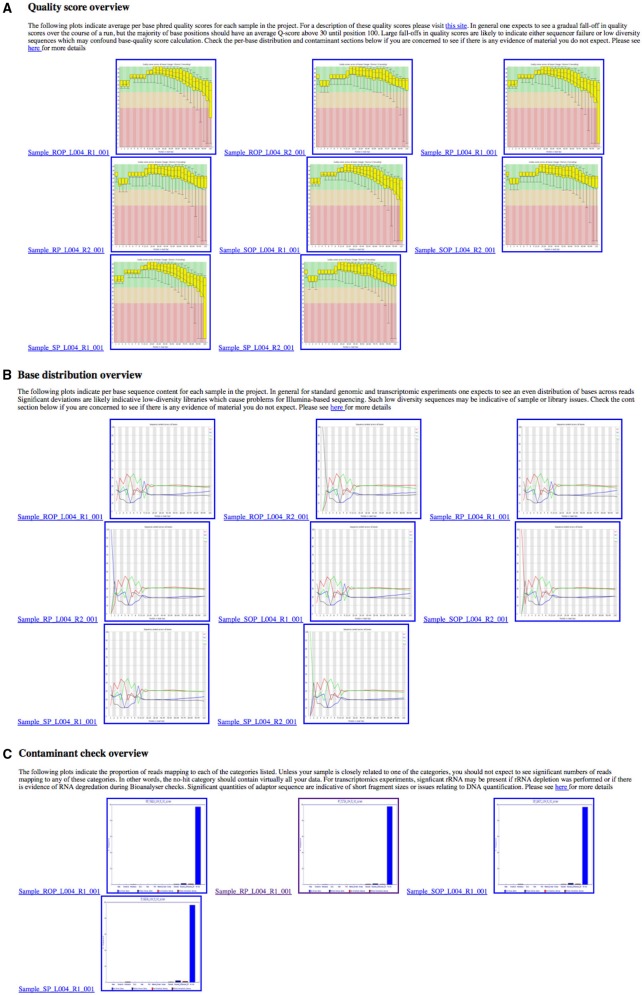
Overview of quality control metrics across multiple samples in a project. These plots are collated into a single HTML summary file for each project, making it easy to see any quality (A), nucleotide (B), or contaminant (C) issues at-a-glance.
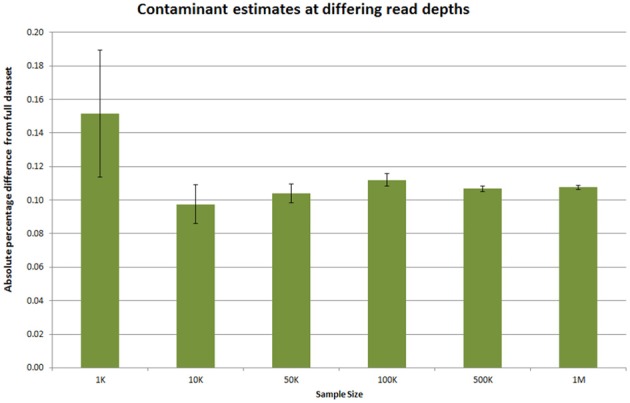
Estimating required read sampling for contaminant checks. Ten million reads from an Illumina RNA-seq dataset was subsampled at various numbers of reads. The number of rRNA contaminant reads in this dataset was 1.86% when calculated over the full dataset. The absolute percentage difference at different sub-sample sizes was calculated for 500 replicates at each depth and the average shown. The error bars indicate the 95% confidence interval for the absolute percentage difference.
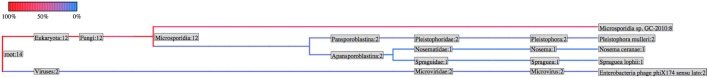
Taxonomy of unmapped reads assembled into contigs. A graphical representation of the number of contigs mapping to each level of the NCBI Taxonomy. The colors represent the number of contigs mapping to each branch.
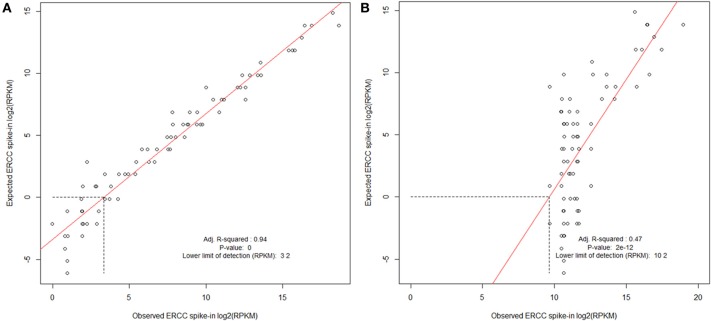
Evaluating ERCC spike-in control results. Examples of cDNA libraries prepared from Arabidopsis thaliana RNA and Mouse infected with Burkholderia pseudomallei RNA containing ERCC Spike-In. Spike-In mix was added to total RNA before preparation of the sequencing library. (A)
A. thaliana poly-A RNA was isolated, and a sequencing library prepared using ScriptSeq v2 (Epicentre). (B) Trial B. pseudomallei sequencing library was prepared from RNA extracted from liver of a B. pseudomallei infected mouse; prokaryotic RNA was enriched using Microbe enrich kit (Invitrogen) and bacterial ribosomal RNA was reduced using MicrobeExpress (Invitrogen) before ScriptSeq v2 sequencing library preparation. ERCC spike in mix is polyadenlyated and the majority would be expected to have been removed during the library preparation resulting in a poor correlation and lower limit of detection, thereby contributing to the protocol development. Libraries were processed and sequenced on the Illumina HiSeq2500. The data were normalized to reads per kilobase of exon model per million mapped reads (RPKM) and filtered using a sensitivity threshold set arbitrarily at 1 RPKM (shown by the horizontal dotted line in at log2 RPKM = 0; Mortazavi et al., 2008).
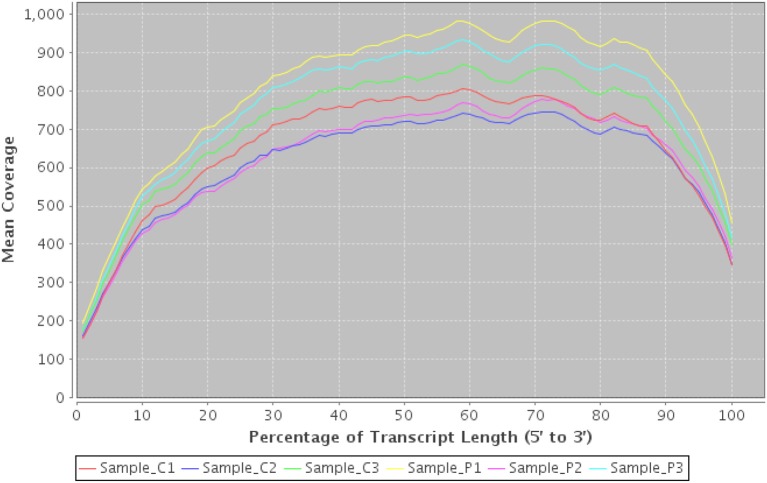
Transcript coverage using RNASeqQC. An example plot from RNASeqQC detailing mean coverage of top 20 transcript abundances for 6 samples. A clear bias can be seen at both 3' and 5' ends which may impact on downstream analysis.
Similar articles
-
Monitoring Error Rates In Illumina Sequencing.J Biomol Tech. 2016 Dec;27(4):125-128. doi: 10.7171/jbt.16-2704-002. Epub 2016 Sep 16. J Biomol Tech. 2016. PMID: 27672352 Free PMC article.
-
AlmostSignificant: simplifying quality control of high-throughput sequencing data.Bioinformatics. 2016 Dec 15;32(24):3850-3851. doi: 10.1093/bioinformatics/btw559. Epub 2016 Aug 24. Bioinformatics. 2016. PMID: 27559158 Free PMC article.
-
Software updates in the Illumina HiSeq platform affect whole-genome bisulfite sequencing.BMC Genomics. 2017 Jan 5;18(1):31. doi: 10.1186/s12864-016-3392-9. BMC Genomics. 2017. PMID: 28056787 Free PMC article.
-
PathoQC: Computationally Efficient Read Preprocessing and Quality Control for High-Throughput Sequencing Data Sets.Cancer Inform. 2015 May 12;13(Suppl 1):167-76. doi: 10.4137/CIN.S13890. eCollection 2014. Cancer Inform. 2015. PMID: 25983538 Free PMC article. Review.
-
Evaluation of next-generation sequencing software in mapping and assembly.J Hum Genet. 2011 Jun;56(6):406-14. doi: 10.1038/jhg.2011.43. Epub 2011 Apr 28. J Hum Genet. 2011. PMID: 21525877 Review.
Cited by
-
Quality assessment and control of high-throughput sequencing data.Front Genet. 2014 Jul 25;5:235. doi: 10.3389/fgene.2014.00235. eCollection 2014. Front Genet. 2014. PMID: 25120556 Free PMC article. No abstract available.
-
Assessment of quality control approaches for metagenomic data analysis.Sci Rep. 2014 Nov 7;4:6957. doi: 10.1038/srep06957. Sci Rep. 2014. PMID: 25376098 Free PMC article.
-
Using QC-Blind for Quality Control and Contamination Screening of Bacteria DNA Sequencing Data Without Reference Genome.Front Microbiol. 2019 Jul 9;10:1560. doi: 10.3389/fmicb.2019.01560. eCollection 2019. Front Microbiol. 2019. PMID: 31354662 Free PMC article.
-
RNA-QC-chain: comprehensive and fast quality control for RNA-Seq data.BMC Genomics. 2018 Feb 14;19(1):144. doi: 10.1186/s12864-018-4503-6. BMC Genomics. 2018. PMID: 29444661 Free PMC article.
-
A Primer on Infectious Disease Bacterial Genomics.Clin Microbiol Rev. 2016 Oct;29(4):881-913. doi: 10.1128/CMR.00001-16. Epub 2016 Sep 7. Clin Microbiol Rev. 2016. PMID: 28590251 Free PMC article. Review.
References
-
- Andrews S. (2010a). FASTQC Package. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Andrews S. (2010b). Fastq-screen Package. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/
-
- Azofeifa D. E., Arguedas H. J., Vargas W. E. (2012) Optical properties of chitin and chitosan biopolymers with application to structural color analysis. Opt. Mater. 35, 175–183 10.1016/j.optmat.2012.07.024 - DOI
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources