

The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis
- PMID: 24904820
- PMCID: PMC4032983
- DOI: 10.3389/fonc.2014.00096
The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis
Abstract
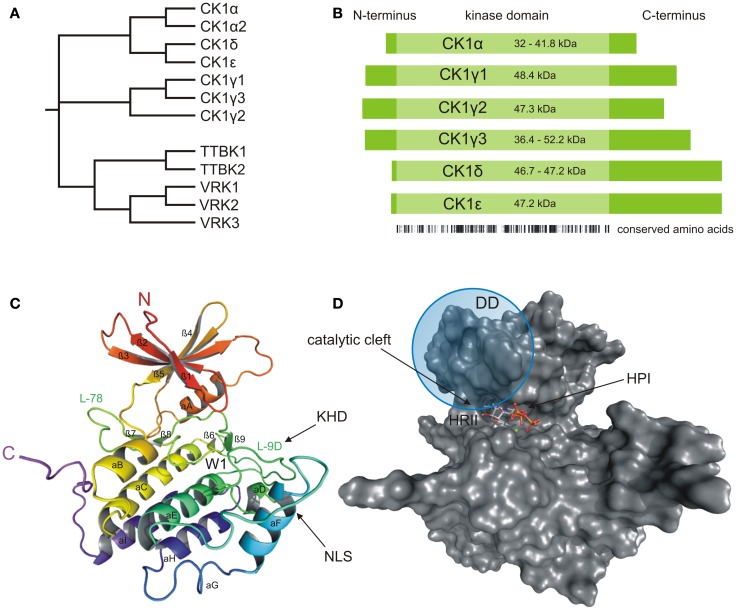
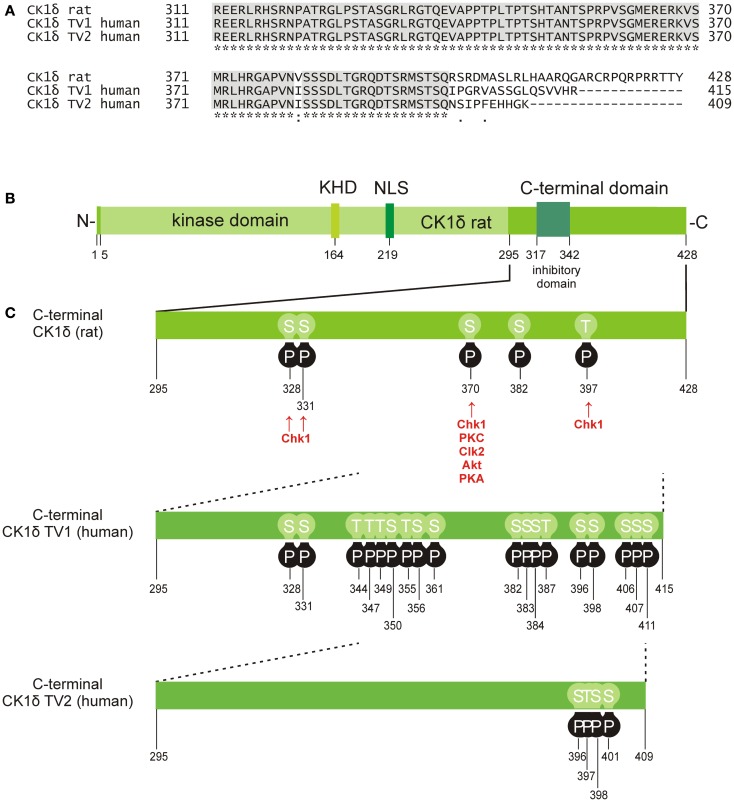
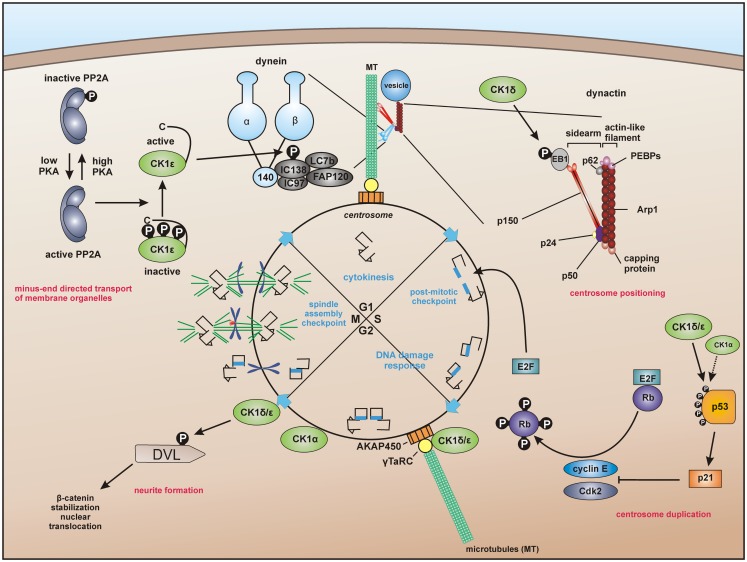
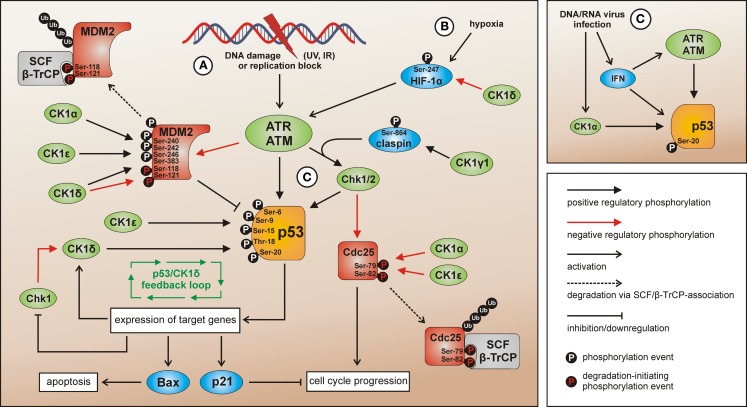
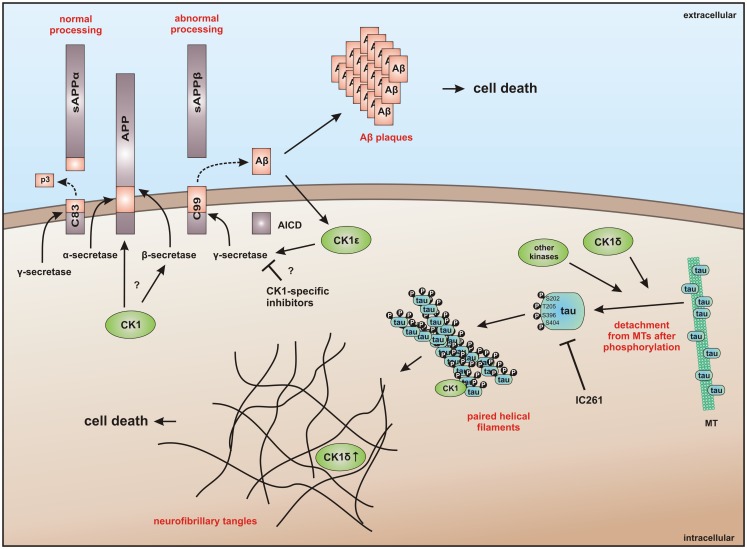
Members of the highly conserved and ubiquitously expressed pleiotropic CK1 family play major regulatory roles in many cellular processes including DNA-processing and repair, proliferation, cytoskeleton dynamics, vesicular trafficking, apoptosis, and cell differentiation. As a consequence of cellular stress conditions, interaction of CK1 with the mitotic spindle is manifold increased pointing to regulatory functions at the mitotic checkpoint. Furthermore, CK1 is able to alter the activity of key proteins in signal transduction and signal integration molecules. In line with this notion, CK1 is tightly connected to the regulation and degradation of β-catenin, p53, and MDM2. Considering the importance of CK1 for accurate cell division and regulation of tumor suppressor functions, it is not surprising that mutations and alterations in the expression and/or activity of CK1 isoforms are often detected in various tumor entities including cancer of the kidney, choriocarcinomas, breast carcinomas, oral cancer, adenocarcinomas of the pancreas, and ovarian cancer. Therefore, scientific effort has enormously increased (i) to understand the regulation of CK1 and its involvement in tumorigenesis- and tumor progression-related signal transduction pathways and (ii) to develop CK1-specific inhibitors for the use in personalized therapy concepts. In this review, we summarize the current knowledge regarding CK1 regulation, function, and interaction with cellular proteins playing central roles in cellular stress-responses and carcinogenesis.
Keywords: casein kinase 1; cellular stress; centrosome; disease; inhibitor; p53; signal transduction; tumorigenesis.
Figures
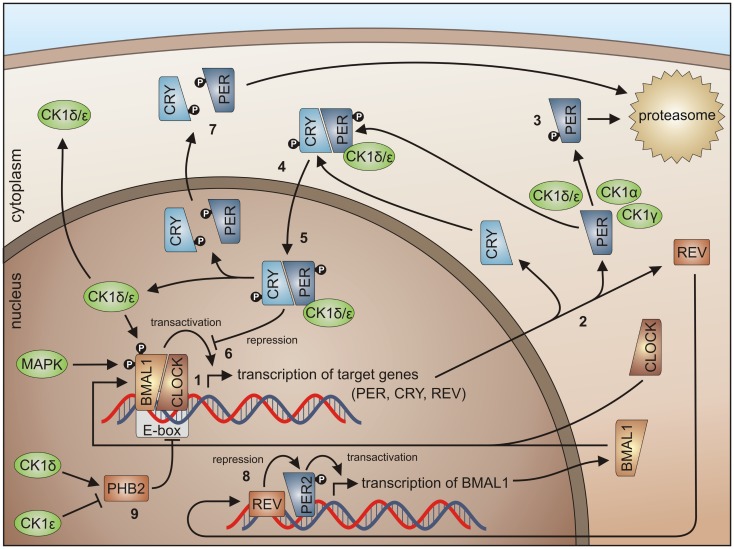
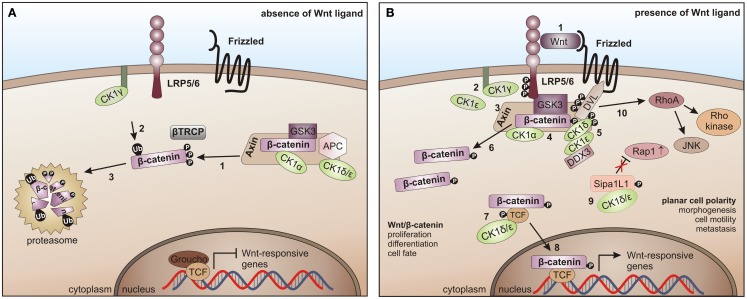
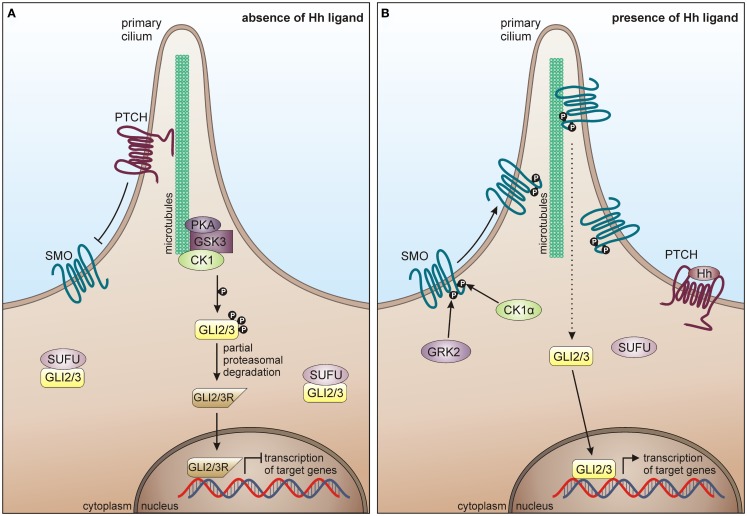
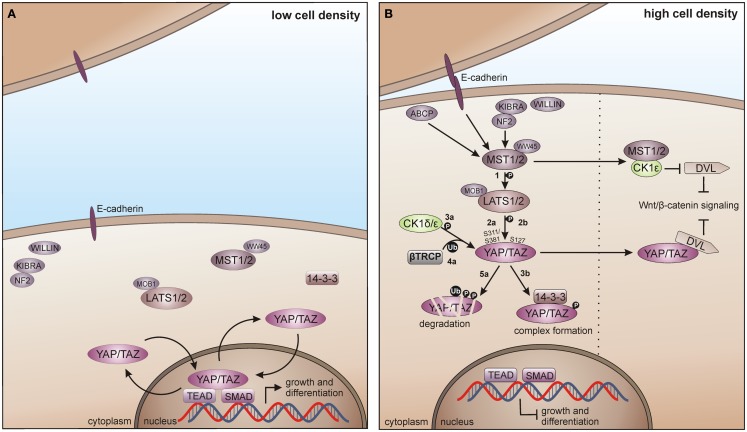
References
-
- Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. Phosphate groups as substrate determinants for casein kinase I action. J Biol Chem (1990) 265(24):14264–9 - PubMed
-
- Flotow H, Roach PJ. Role of acidic residues as substrate determinants for casein kinase I. J Biol Chem (1991) 266(6):3724–7 - PubMed
-
- Graves PR, Haas DW, Hagedorn CH, DePaoli-Roach AA, Roach PJ. Molecular cloning, expression, and characterization of a 49-kilodalton casein kinase I isoform from rat testis. J Biol Chem (1993) 268(9):6394–401 - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous