

Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer
- PMID: 24904834
- PMCID: PMC4033620
- DOI: 10.3389/fonc.2014.00123
Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer
Abstract
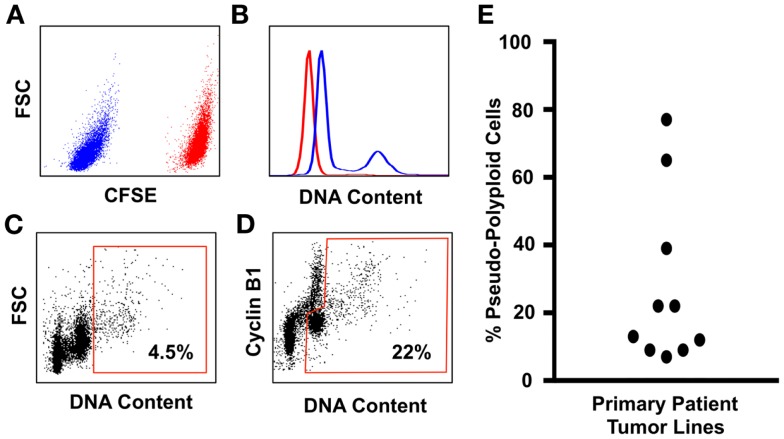
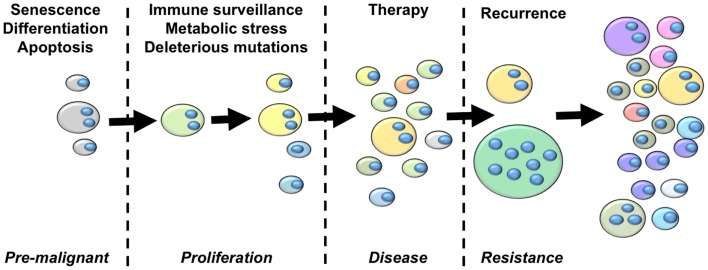
Tumor evolution presents a formidable obstacle that currently prevents the development of truly curative treatments for cancer. In this perspective, we advocate for the hypothesis that tumor cells with significantly elevated genomic content (polyploid tumor cells) facilitate rapid tumor evolution and the acquisition of therapy resistance in multiple incurable cancers. We appeal to studies conducted in yeast, cancer models, and cancer patients, which all converge on the hypothesis that polyploidy enables large phenotypic leaps, providing access to many different therapy-resistant phenotypes. We develop a flow-cytometry based method for quantifying the prevalence of polyploid tumor cells, and show the frequency of these cells in patient tumors may be higher than is generally appreciated. We then present recent studies identifying promising new therapeutic strategies that could be used to specifically target polyploid tumor cells in cancer patients. We argue that these therapeutic approaches should be incorporated into new treatment strategies aimed at blocking tumor evolution by killing the highly evolvable, therapy-resistant polyploid cell subpopulations, thus helping to maintain patient tumors in a drug sensitive state.
Keywords: aneuploidy; cancer stem cell; chromosomal instability; hyperdiploidy; polyploidy; therapy resistance; tumor evolution; tumor initiation.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases