

Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking
- PMID: 24905460
- PMCID: PMC4203550
- DOI: 10.4161/auto.28912
Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking
Abstract
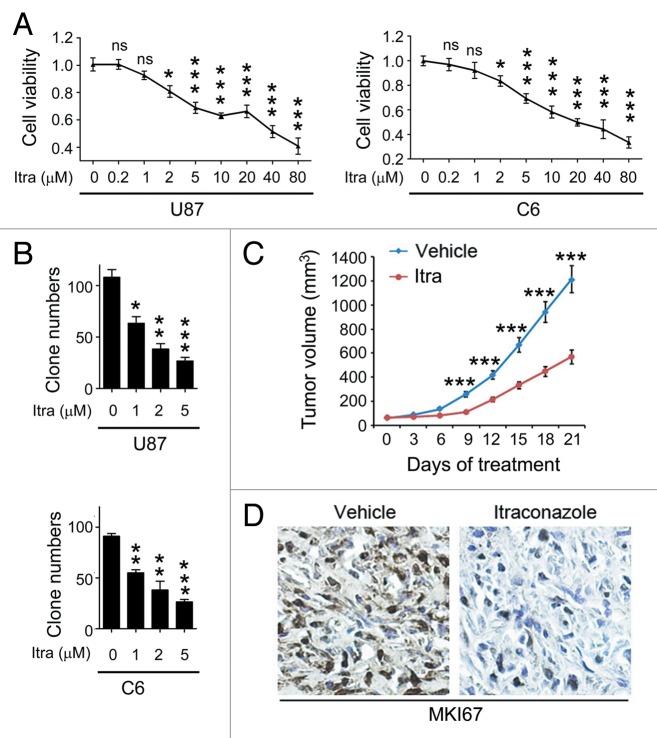
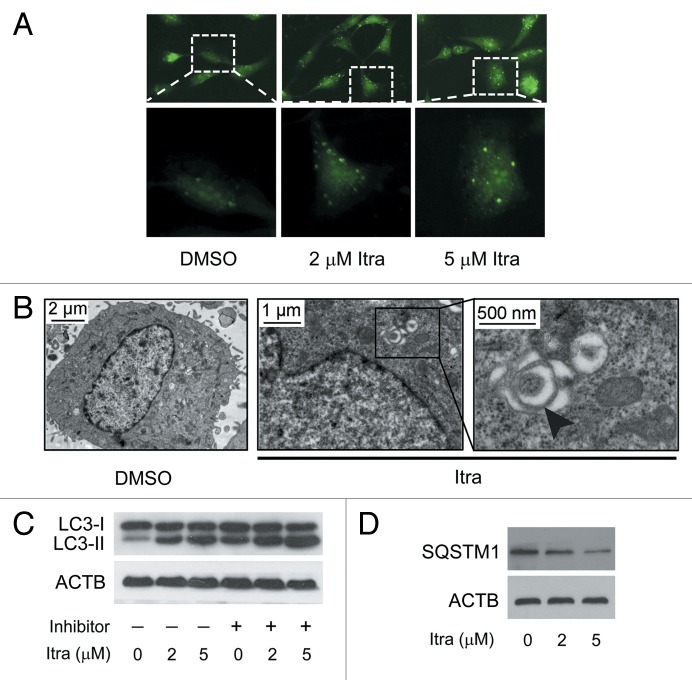
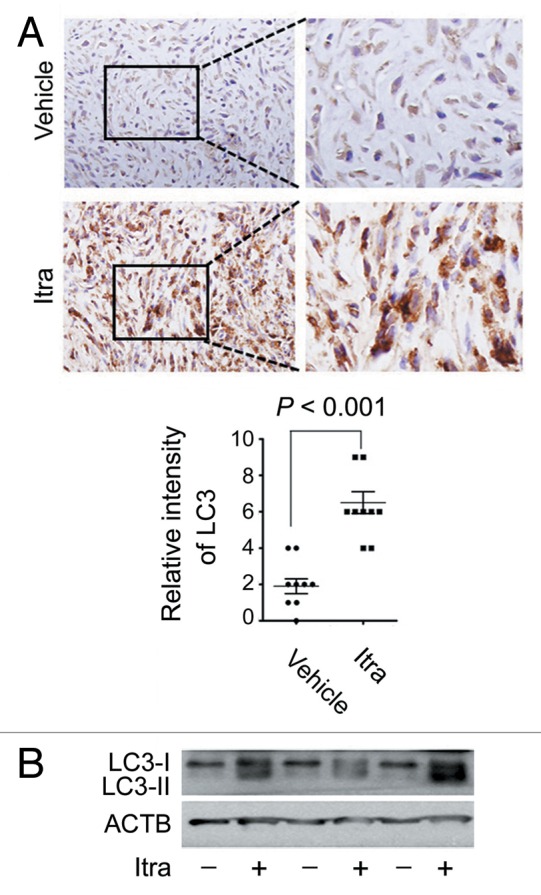
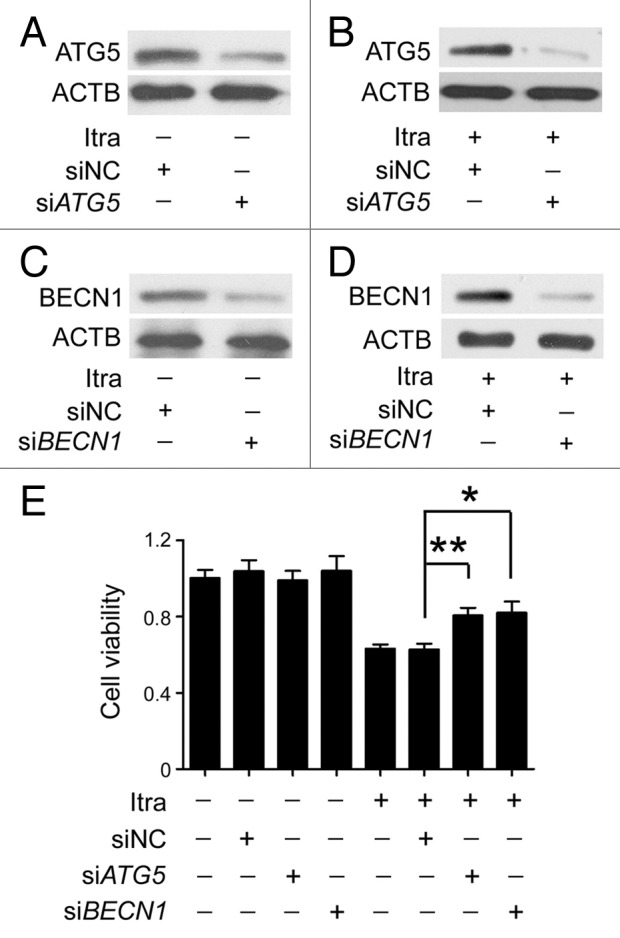
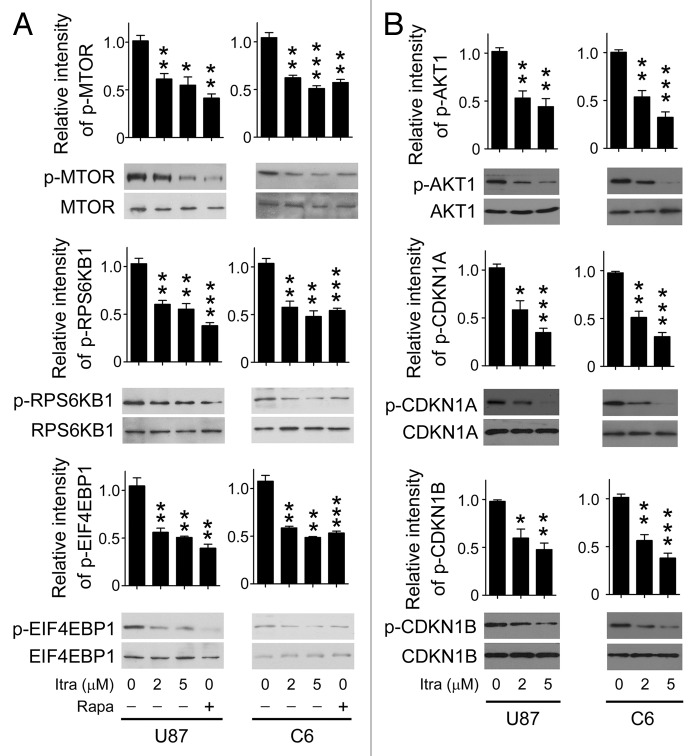
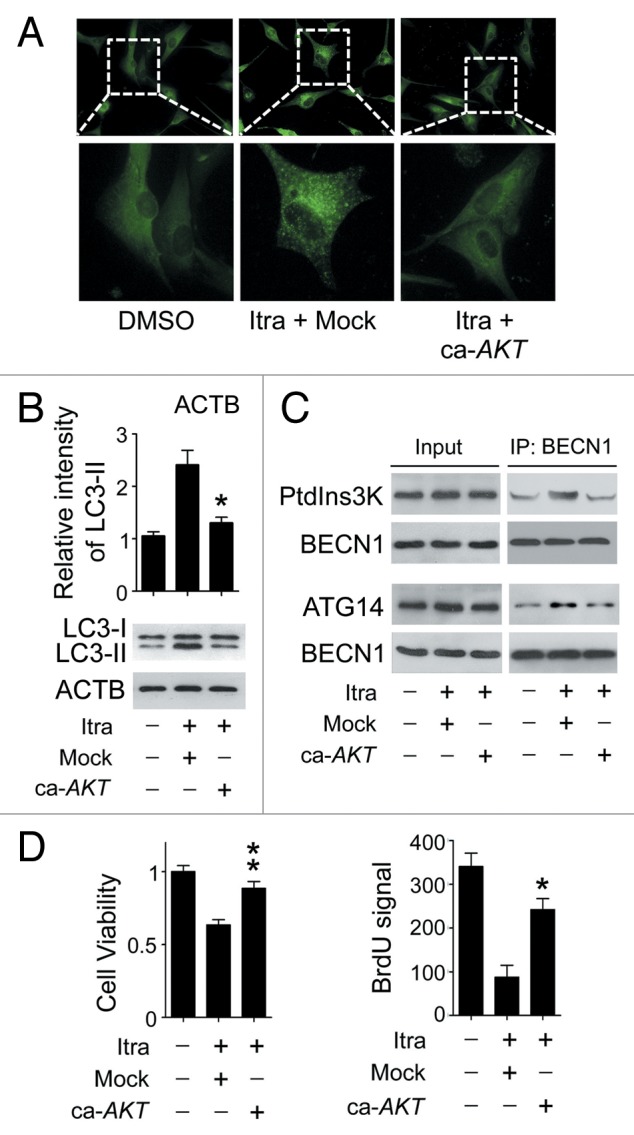
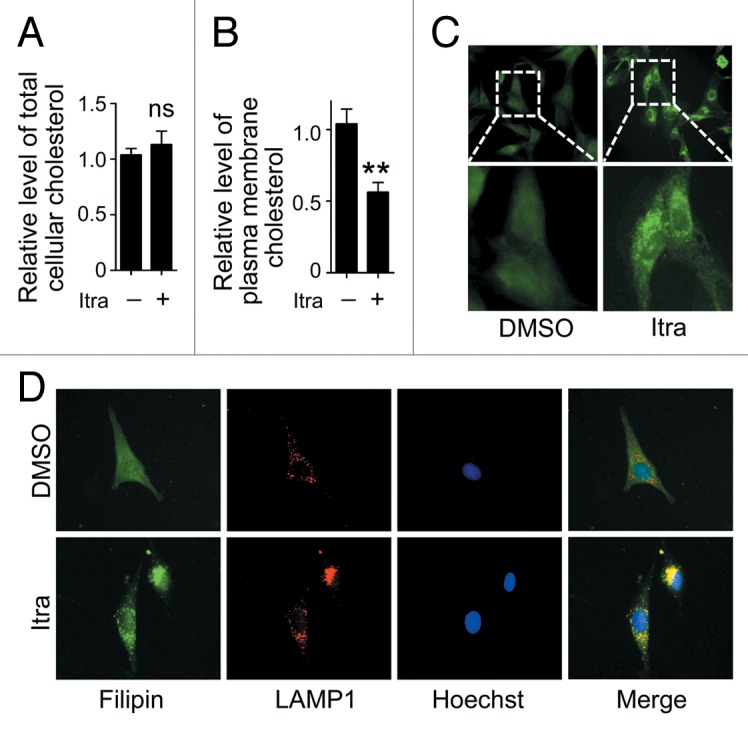
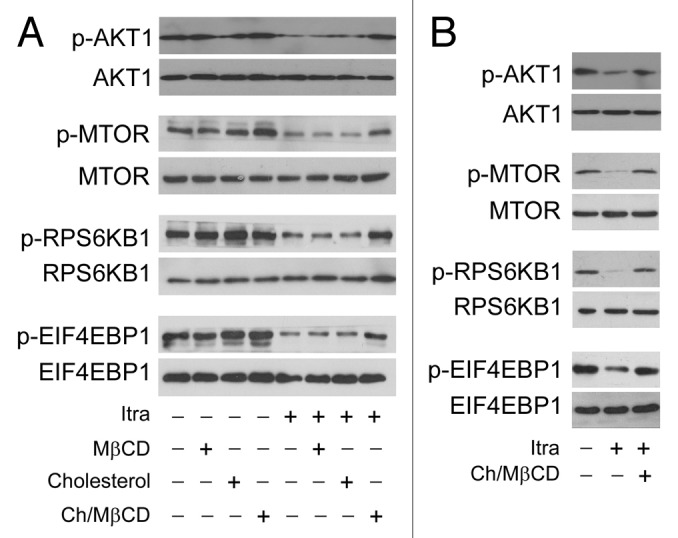
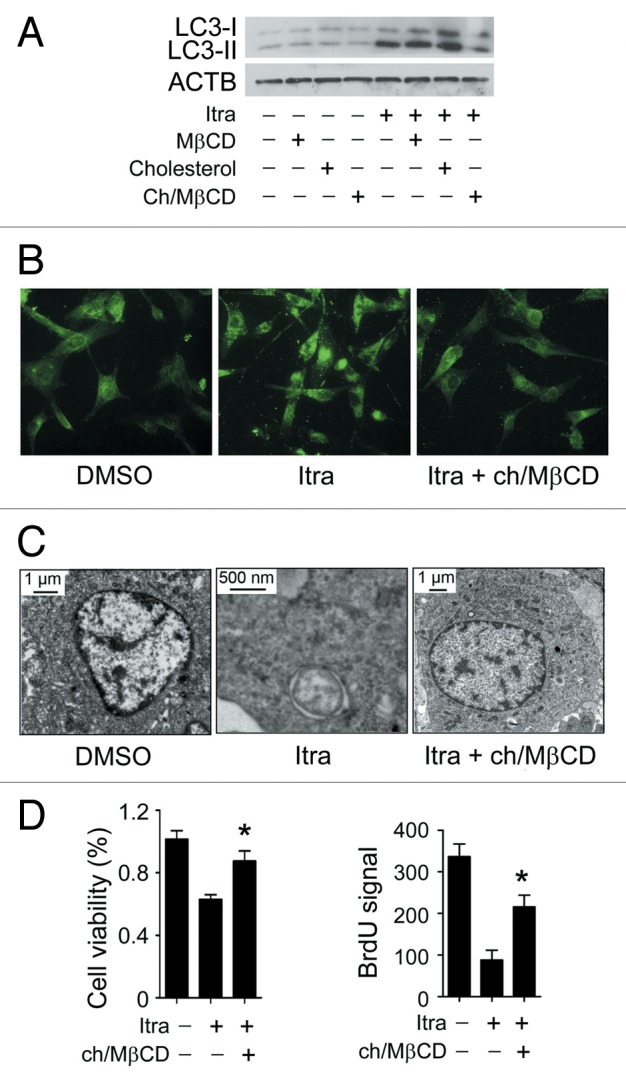
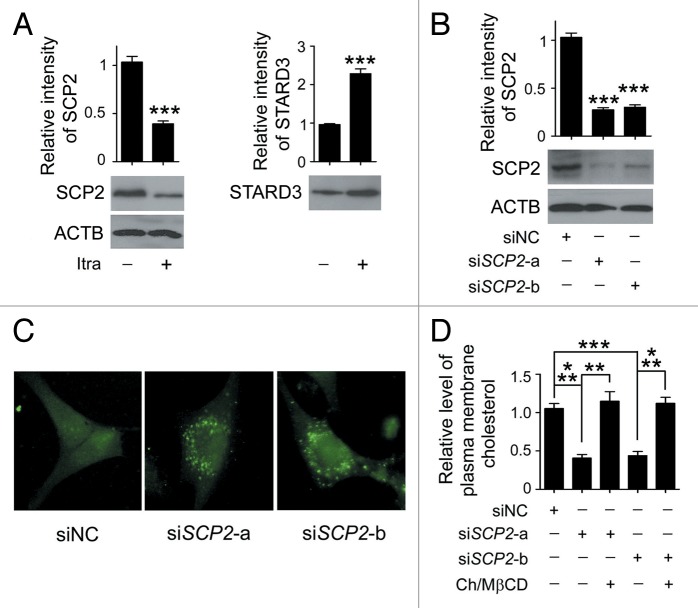
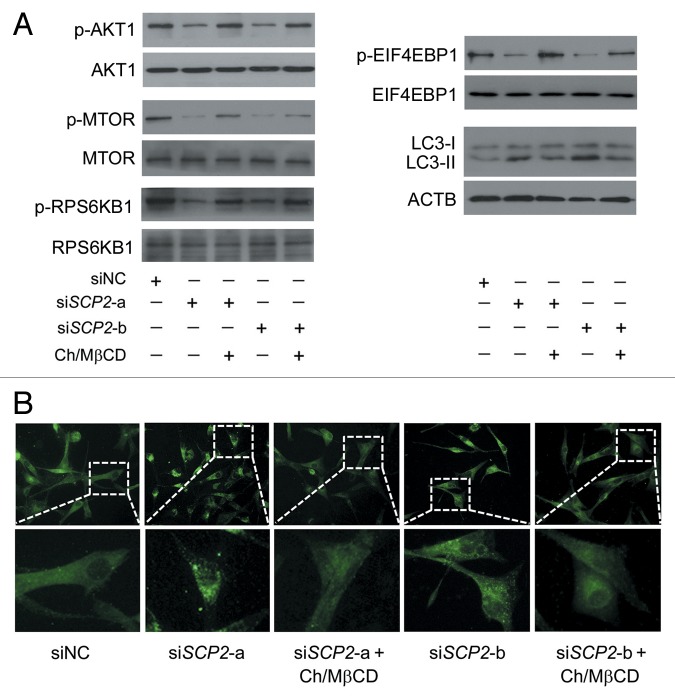
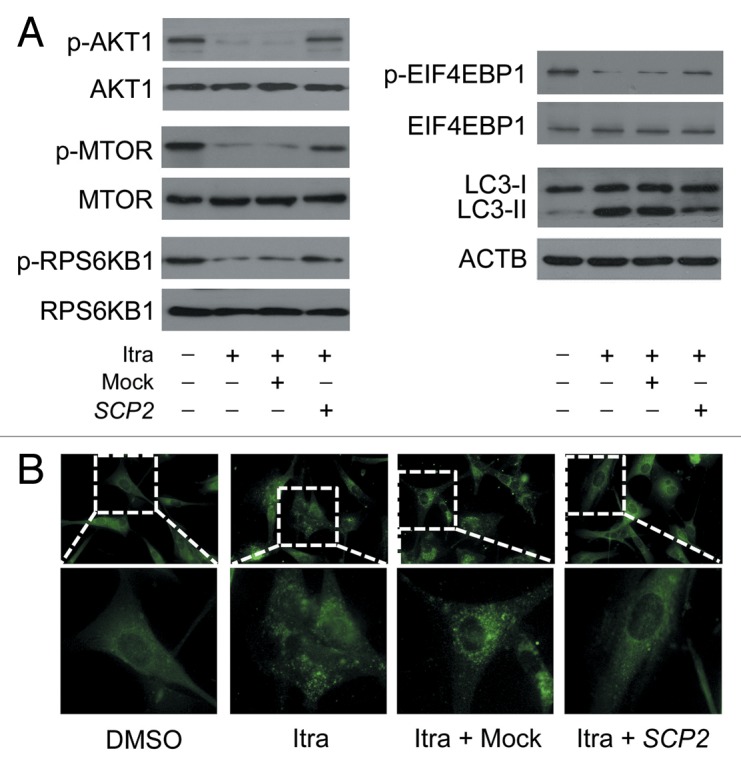
Glioblastoma is one of the most aggressive human cancers with poor prognosis, and therefore a critical need exists for novel therapeutic strategies for management of glioblastoma patients. Itraconazole, a traditional antifungal drug, has been identified as a novel potential anticancer agent due to its inhibitory effects on cell proliferation and tumor angiogenesis; however, the molecular mechanisms involved are still unclear. Here, we show that itraconazole inhibits the proliferation of glioblastoma cells both in vitro and in vivo. Notably, we demonstrate that treatment with itraconazole induces autophagic progression in glioblastoma cells, while blockage of autophagy markedly reverses the antiproliferative activities of itraconazole, suggesting an antitumor effect of autophagy in response to itraconazole treatment. Functional studies revealed that itraconazole retarded the trafficking of cholesterol from late endosomes and lysosomes to the plasma membrane by reducing the levels of SCP2, resulting in repression of AKT1-MTOR signaling, induction of autophagy, and finally inhibition of cell proliferation. Together, our studies provide new insights into the molecular mechanisms regarding the antitumor activities of itraconazole, and may further assist both the pharmacological investigation and rational use of itraconazole in potential clinical applications.
Keywords: BECN1-PtdIns3K complex; antiproliferative; autophagy; cholesterol trafficking; itraconazole.
Figures
Similar articles
-
Ganoderic acid A holds promising cytotoxicity on human glioblastoma mediated by incurring apoptosis and autophagy and inactivating PI3K/AKT signaling pathway.J Biochem Mol Toxicol. 2019 Nov;33(11):e22392. doi: 10.1002/jbt.22392. Epub 2019 Sep 10. J Biochem Mol Toxicol. 2019. PMID: 31503386
-
Oncogenic Ras is downregulated by ARHI and induces autophagy by Ras/AKT/mTOR pathway in glioblastoma.BMC Cancer. 2019 May 14;19(1):441. doi: 10.1186/s12885-019-5643-z. BMC Cancer. 2019. PMID: 31088402 Free PMC article.
-
Simultaneous Targeting of NPC1 and VDAC1 by Itraconazole Leads to Synergistic Inhibition of mTOR Signaling and Angiogenesis.ACS Chem Biol. 2017 Jan 20;12(1):174-182. doi: 10.1021/acschembio.6b00849. Epub 2016 Dec 2. ACS Chem Biol. 2017. PMID: 28103683 Free PMC article.
-
Wnt/beta-catenin and PI3K/Akt/mTOR Signaling Pathways in Glioblastoma: Two Main Targets for Drug Design: A Review.Curr Pharm Des. 2020;26(15):1729-1741. doi: 10.2174/1381612826666200131100630. Curr Pharm Des. 2020. PMID: 32003685 Review.
-
PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma.Oncotarget. 2016 May 31;7(22):33440-50. doi: 10.18632/oncotarget.7961. Oncotarget. 2016. PMID: 26967052 Free PMC article. Review.
Cited by
-
A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials.Cancers (Basel). 2021 Apr 9;13(8):1795. doi: 10.3390/cancers13081795. Cancers (Basel). 2021. PMID: 33918704 Free PMC article. Review.
-
Non-self glycan structures as possible modulators of cancer progression: would polysaccharides from Cryptococcus spp. impact this phenomenon?Braz J Microbiol. 2023 Jun;54(2):907-919. doi: 10.1007/s42770-023-00936-0. Epub 2023 Feb 25. Braz J Microbiol. 2023. PMID: 36840821 Free PMC article. Review.
-
Sphingolipid biosynthetic pathway is crucial for growth, biofilm formation and membrane integrity of Scedosporium boydii.Future Med Chem. 2019 Nov;11(22):2905-2917. doi: 10.4155/fmc-2019-0186. Epub 2019 Nov 12. Future Med Chem. 2019. PMID: 31713454 Free PMC article.
-
Cholesterol metabolism and its implication in glioblastoma therapy.J Cancer. 2022 Mar 14;13(6):1745-1757. doi: 10.7150/jca.63609. eCollection 2022. J Cancer. 2022. PMID: 35399707 Free PMC article. Review.
-
Berberine ameliorates blockade of autophagic flux in the liver by regulating cholesterol metabolism and inhibiting COX2-prostaglandin synthesis.Cell Death Dis. 2018 Aug 1;9(8):824. doi: 10.1038/s41419-018-0890-5. Cell Death Dis. 2018. PMID: 30068904 Free PMC article.
References
-
- Lima FR, Kahn SA, Soletti RC, Biasoli D, Alves T, da Fonseca AC, Garcia C, Romão L, Brito J, Holanda-Afonso R, et al. Glioblastoma: therapeutic challenges, what lies ahead. Biochim Biophys Acta. 2012;1826:338–49. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous