

Mechanisms of acetaminophen-induced cell death in primary human hepatocytes
- PMID: 24905542
- PMCID: PMC4171351
- DOI: 10.1016/j.taap.2014.05.010
Mechanisms of acetaminophen-induced cell death in primary human hepatocytes
Abstract
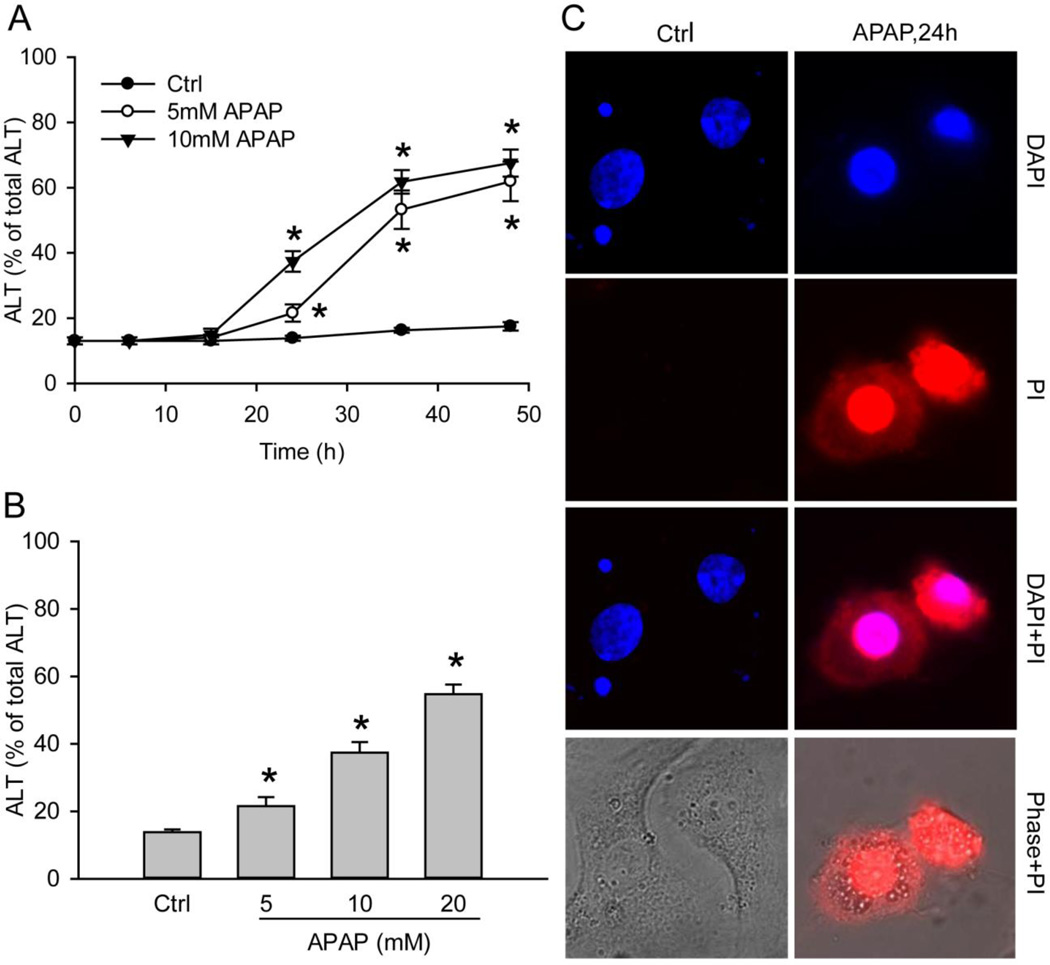
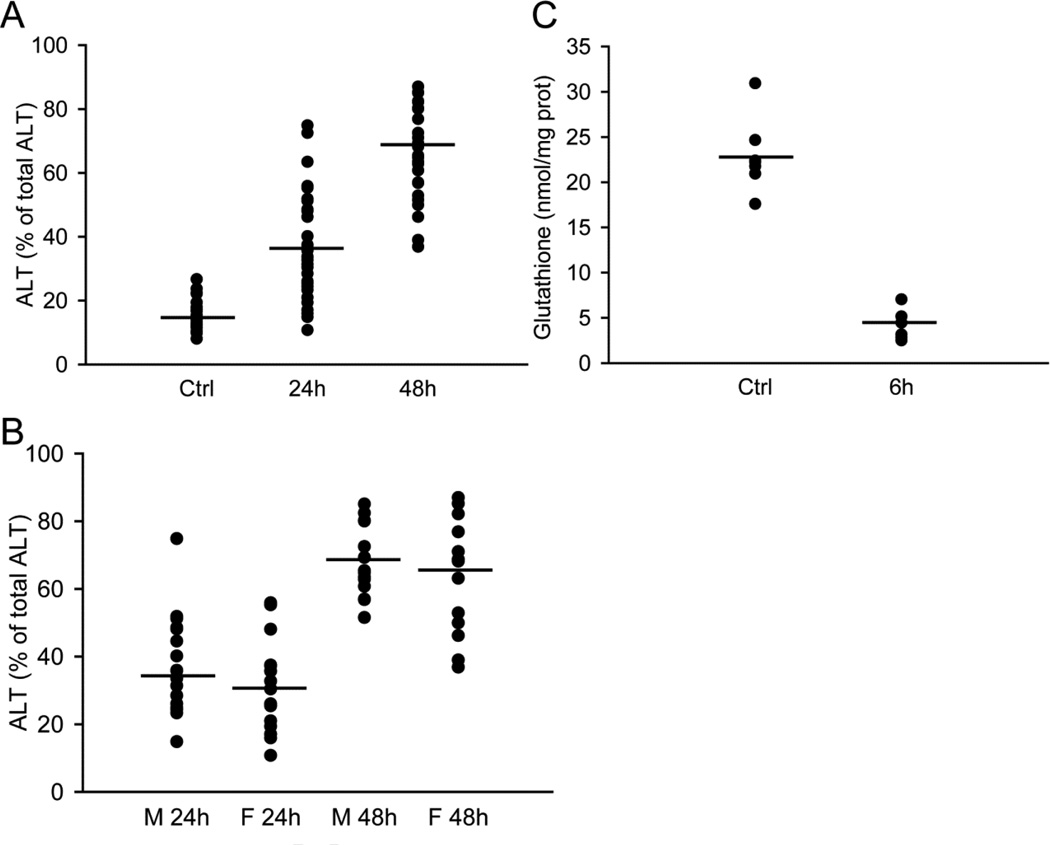
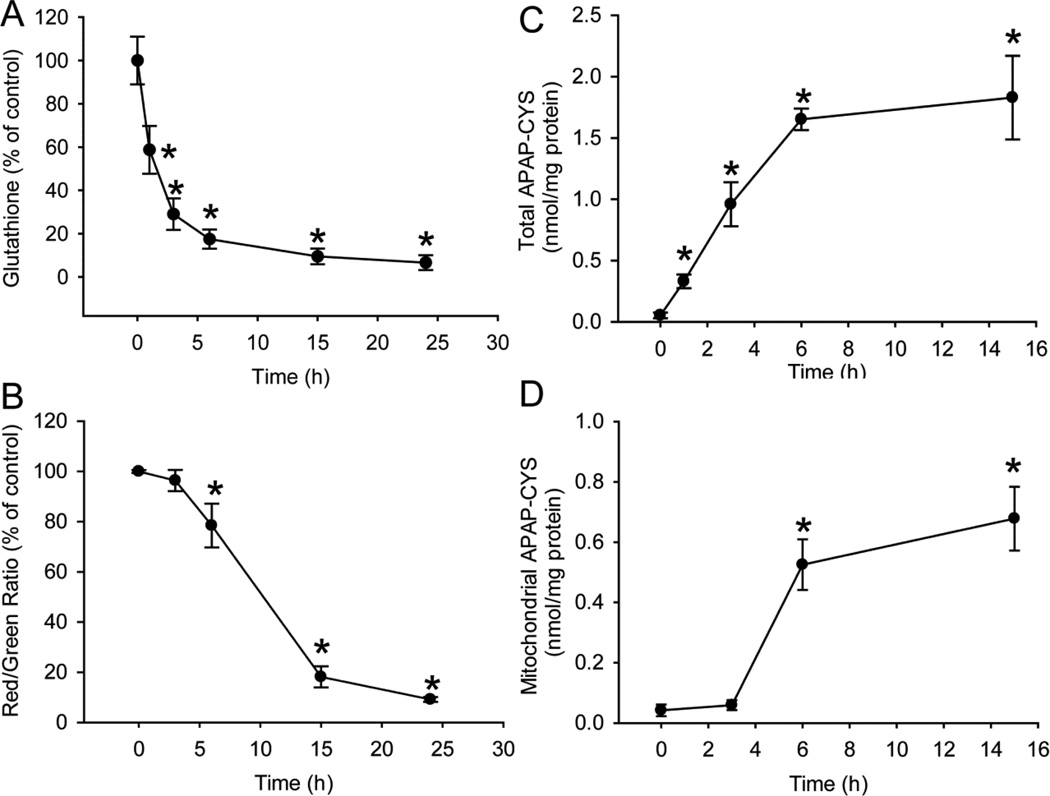
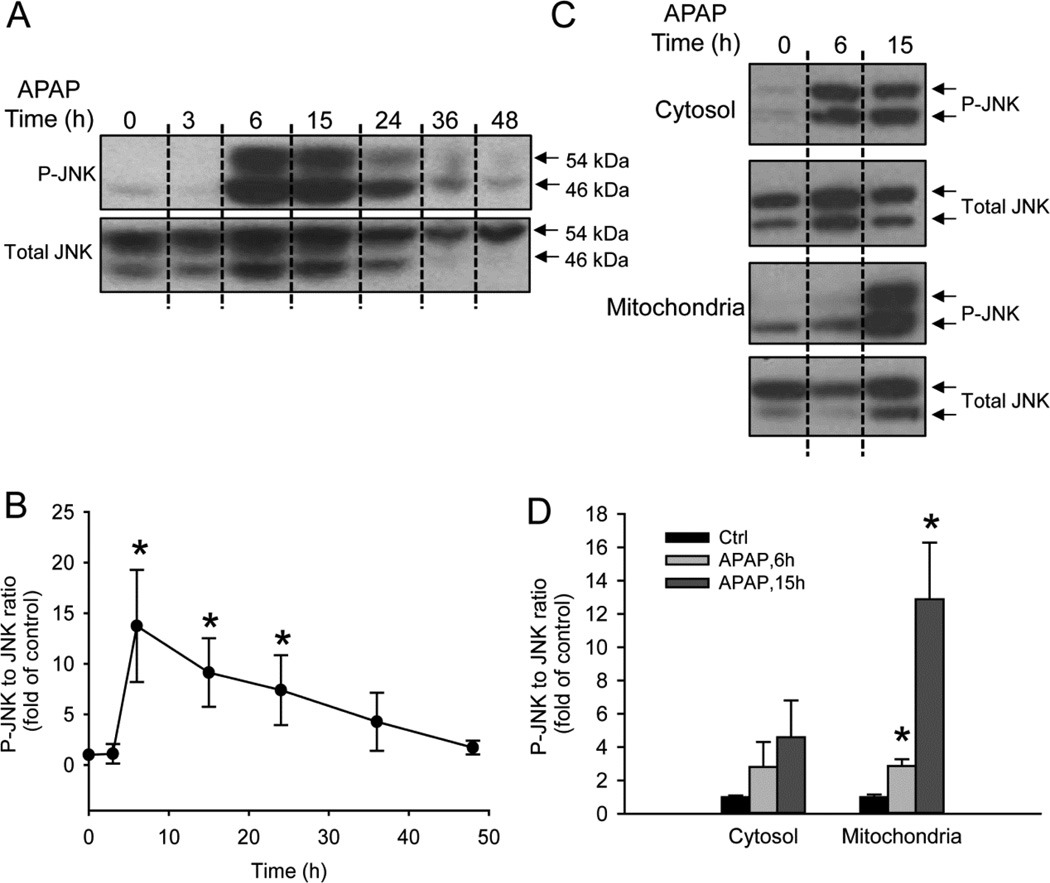
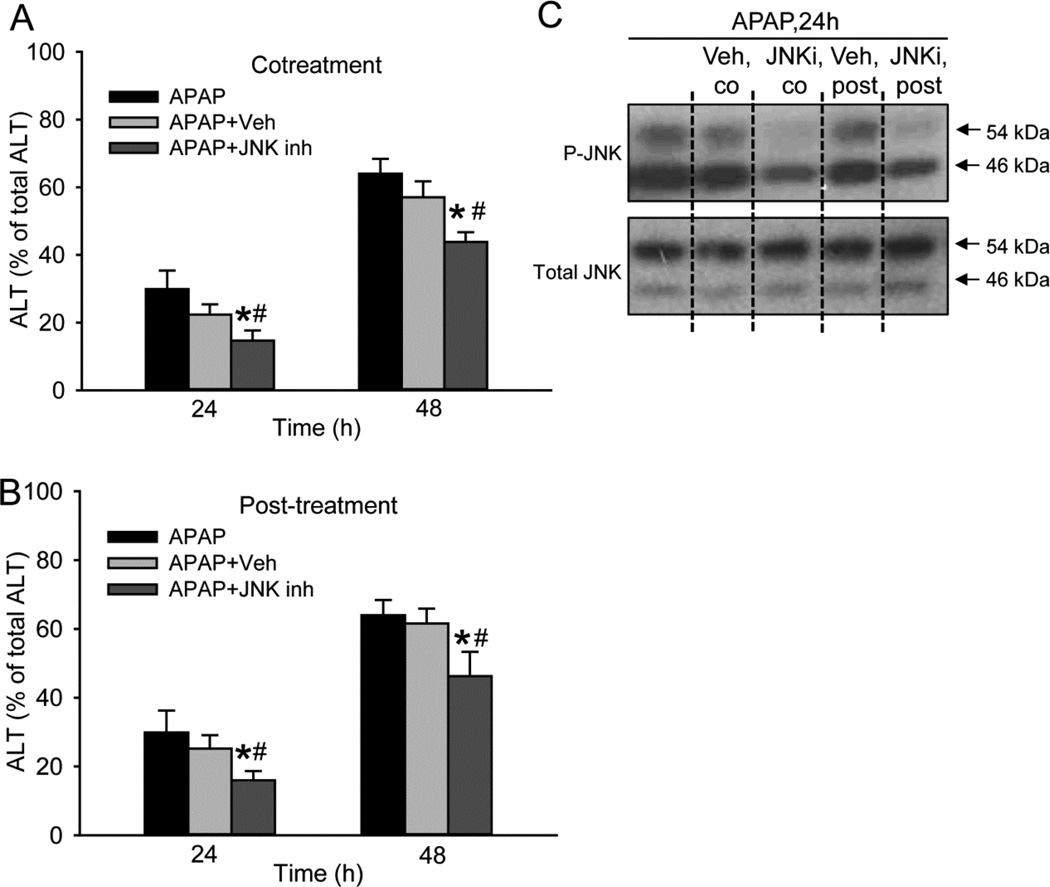
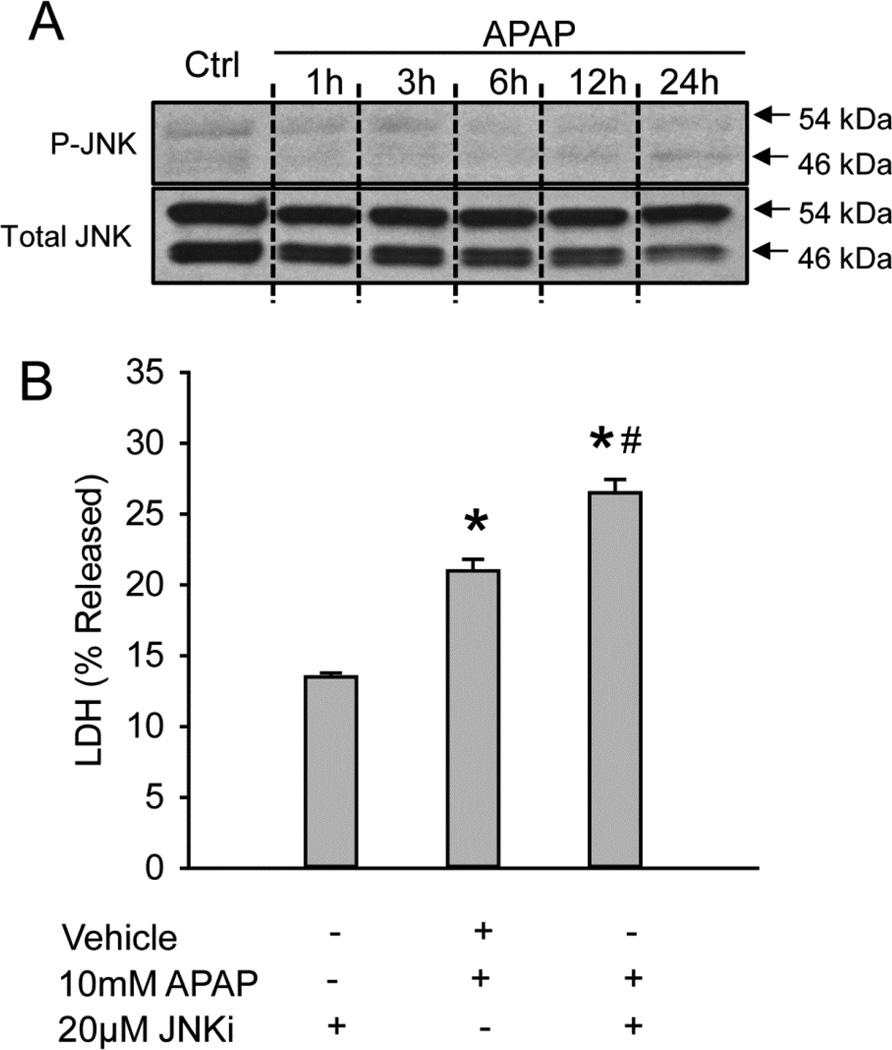
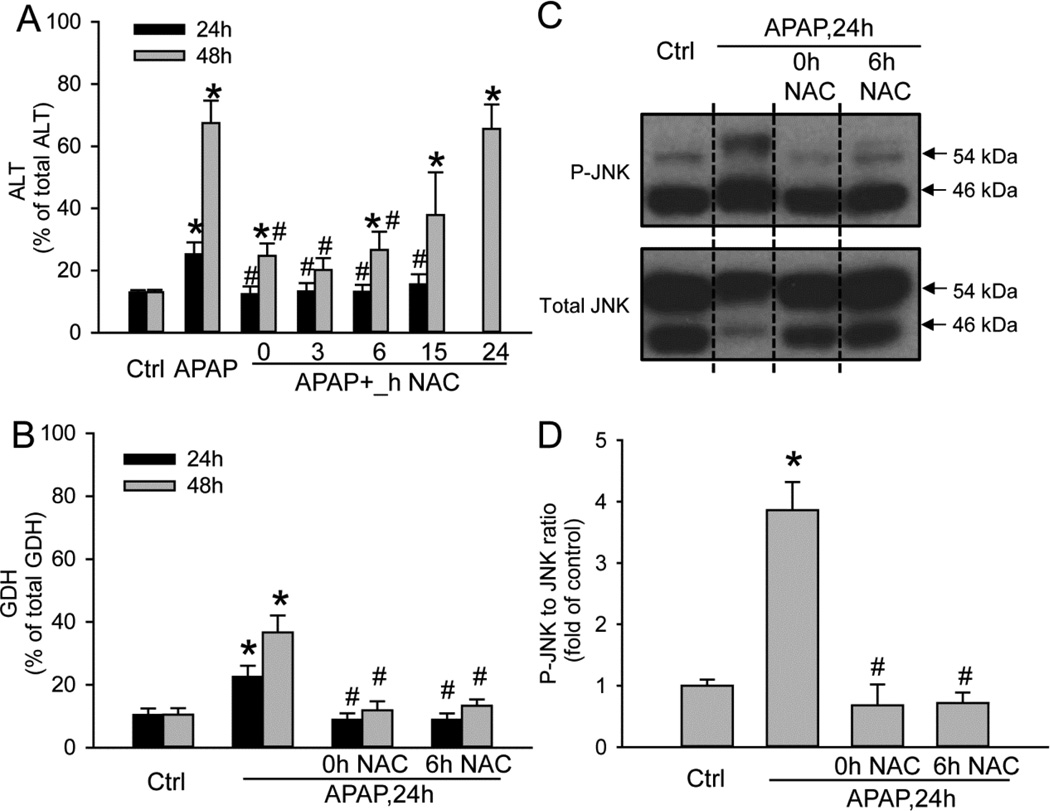
Acetaminophen (APAP) overdose is the most prevalent cause of drug-induced liver injury in western countries. Numerous studies have been conducted to investigate the mechanisms of injury after APAP overdose in various animal models; however, the importance of these mechanisms for humans remains unclear. Here we investigated APAP hepatotoxicity using freshly isolated primary human hepatocytes (PHH) from either donor livers or liver resections. PHH were exposed to 5mM, 10mM or 20mM APAP over a period of 48 h and multiple parameters were assessed. APAP dose-dependently induced significant hepatocyte necrosis starting from 24h, which correlated with the clinical onset of human liver injury after APAP overdose. Interestingly, cellular glutathione was depleted rapidly during the first 3h. APAP also resulted in early formation of APAP-protein adducts (measured in whole cell lysate and in mitochondria) and mitochondrial dysfunction, indicated by the loss of mitochondrial membrane potential after 12h. Furthermore, APAP time-dependently triggered c-Jun N-terminal kinase (JNK) activation in the cytosol and translocation of phospho-JNK to the mitochondria. Both co-treatment and post-treatment (3h) with the JNK inhibitor SP600125 reduced JNK activation and significantly attenuated cell death at 24h and 48h after APAP. The clinical antidote N-acetylcysteine offered almost complete protection even if administered 6h after APAP and a partial protection when given at 15 h.
Conclusion: These data highlight important mechanistic events in APAP toxicity in PHH and indicate a critical role of JNK in the progression of injury after APAP in humans. The JNK pathway may represent a therapeutic target in the clinic.
Keywords: Acetaminophen protein adducts; Drug-induced liver injury; Mitochondrial dysfunction; Oncotic necrosis; c-Jun-N-terminal kinase.
Copyright © 2014 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol. 2012;56:1070–1079. - PMC - PubMed
-
- Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci. 2004;80:343–349. - PubMed
-
- Corcoran GB, Racz WJ, Smith CV, Mitchell JR. Effects of N-acetylcysteine on acetaminophen covalent binding and hepatic necrosis in mice. J Pharmacol Exp Ther. 1985;232:864–872. - PubMed
-
- Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2005;315:879–887. - PubMed
-
- Davern TJ2nd, James LP, Hinson JA, Polson J, Larson AM, Fontana RJ, Lalani E, Munoz S, Shakil AO, Lee WM Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687–694. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
