

Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells
- PMID: 24907273
- PMCID: PMC4118095
- DOI: 10.1074/jbc.M114.564625
Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells
Erratum in
-
Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells.J Biol Chem. 2017 Feb 10;292(6):2556. doi: 10.1074/jbc.A114.564625. J Biol Chem. 2017. PMID: 28188222 Free PMC article. No abstract available.
Abstract
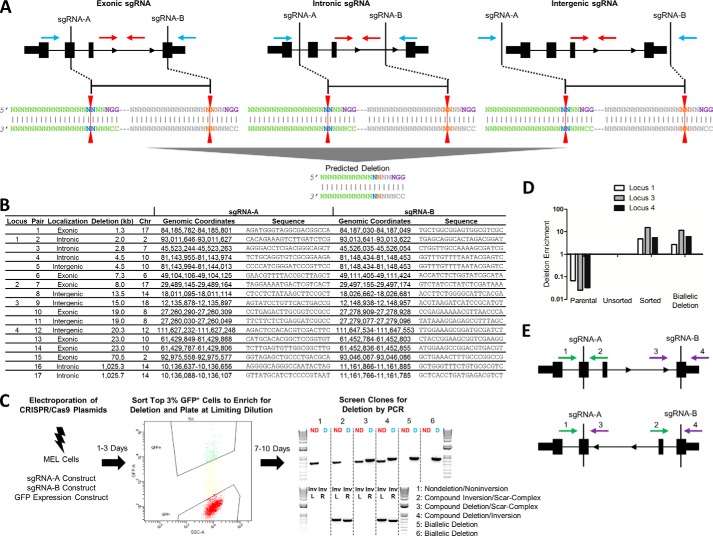
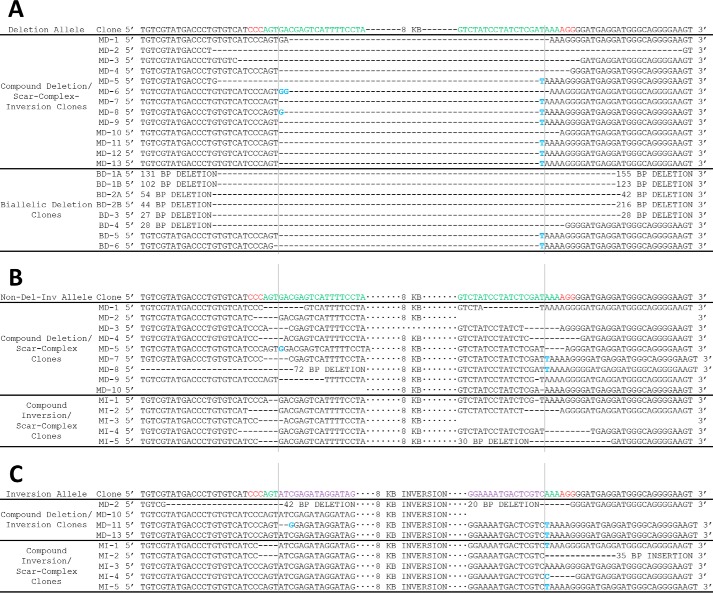
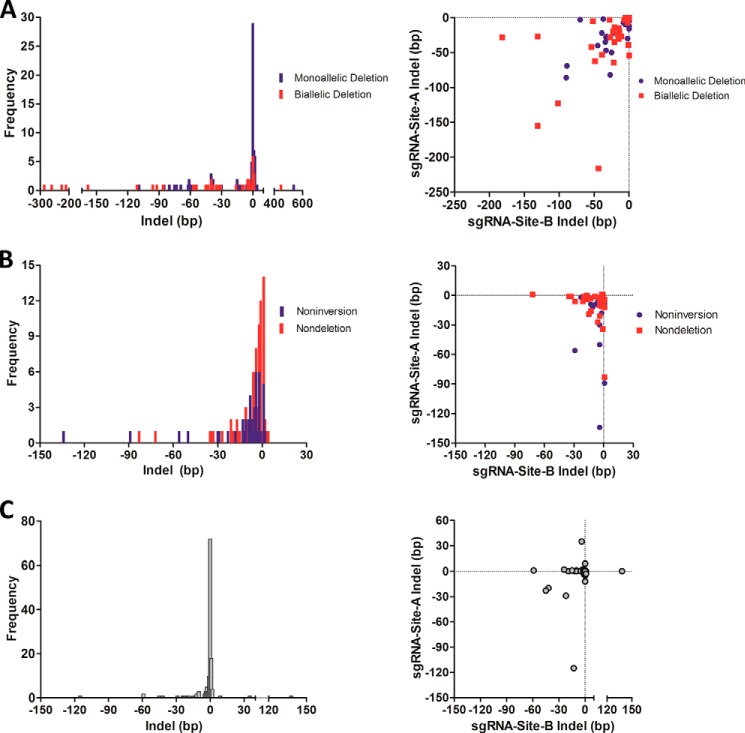
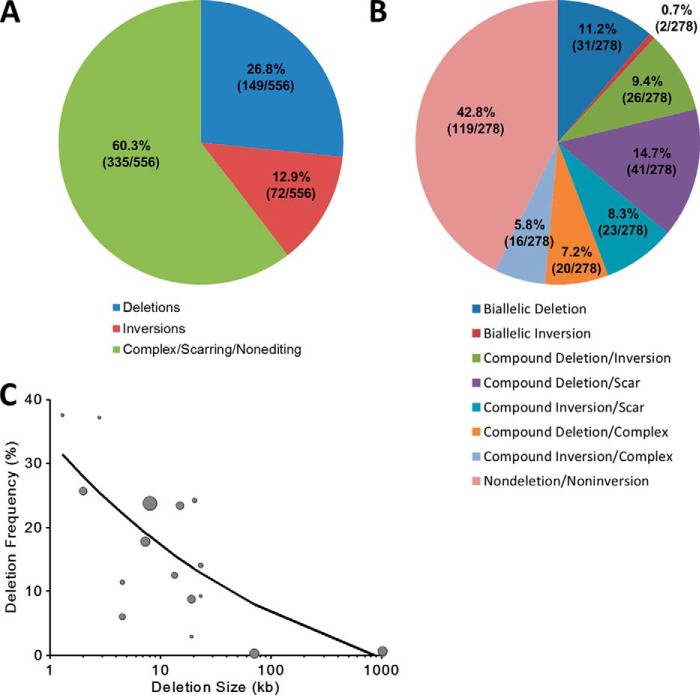
The clustered regularly interspaced short [corrected] palindromic repeats (CRISPR)/CRISPR-associated (Cas) 9 nuclease system has provided a powerful tool for genome engineering. Double strand breaks may trigger nonhomologous end joining repair, leading to frameshift mutations, or homology-directed repair using an extrachromosomal template. Alternatively, genomic deletions may be produced by a pair of double strand breaks. The efficiency of CRISPR/Cas9-mediated genomic deletions has not been systematically explored. Here, we present a methodology for the production of deletions in mammalian cells, ranging from 1.3 kb to greater than 1 Mb. We observed a high frequency of intended genomic deletions. Nondeleted alleles are nonetheless often edited with inversions or small insertion/deletions produced at CRISPR recognition sites. Deleted alleles also typically include small insertion/deletions at predicted deletion junctions. We retrieved cells with biallelic deletion at a frequency exceeding that of probabilistic expectation. We demonstrate an inverse relationship between deletion frequency and deletion size. This work suggests that CRISPR/Cas9 is a robust system to produce a spectrum of genomic deletions to allow investigation of genes and genetic elements.
Keywords: CRISPR; Cas9; Gene Expression; Gene Knock-out; Gene Regulation; Genetics; Genome Engineering; Genomic Deletion; Genomics.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures



References
-
- Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D. A., Horvath P. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 - PubMed
Publication types
MeSH terms
Grants and funding
- R01 HL032259/HL/NHLBI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P30 DK049216/DK/NIDDK NIH HHS/United States
- P01HL032262/HL/NHLBI NIH HHS/United States
- R01DK070838/DK/NIDDK NIH HHS/United States
- T32HL007574/HL/NHLBI NIH HHS/United States
- F32DK098866/DK/NIDDK NIH HHS/United States
- R01HL032259/HL/NHLBI NIH HHS/United States
- R01 AI084905/AI/NIAID NIH HHS/United States
- K08DK093705/DK/NIDDK NIH HHS/United States
- R01AI084905/AI/NIAID NIH HHS/United States
- T32 HL007574/HL/NHLBI NIH HHS/United States
- K08 DK093705/DK/NIDDK NIH HHS/United States
- P01 HL032262/HL/NHLBI NIH HHS/United States
- R01 DK070838/DK/NIDDK NIH HHS/United States
- F32 DK098866/DK/NIDDK NIH HHS/United States
- P30DK049216/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
