

ABRA: improved coding indel detection via assembly-based realignment
- PMID: 24907369
- PMCID: PMC4173014
- DOI: 10.1093/bioinformatics/btu376
ABRA: improved coding indel detection via assembly-based realignment
Abstract
Motivation: Variant detection from next-generation sequencing (NGS) data is an increasingly vital aspect of disease diagnosis, treatment and research. Commonly used NGS-variant analysis tools generally rely on accurately mapped short reads to identify somatic variants and germ-line genotypes. Existing NGS read mappers have difficulty accurately mapping short reads containing complex variation (i.e. more than a single base change), thus making identification of such variants difficult or impossible. Insertions and deletions (indels) in particular have been an area of great difficulty. Indels are frequent and can have substantial impact on function, which makes their detection all the more imperative.
Results: We present ABRA, an assembly-based realigner, which uses an efficient and flexible localized de novo assembly followed by global realignment to more accurately remap reads. This results in enhanced performance for indel detection as well as improved accuracy in variant allele frequency estimation.
Availability and implementation: ABRA is implemented in a combination of Java and C/C++ and is freely available for download at https://github.com/mozack/abra.
© The Author 2014. Published by Oxford University Press.
Figures
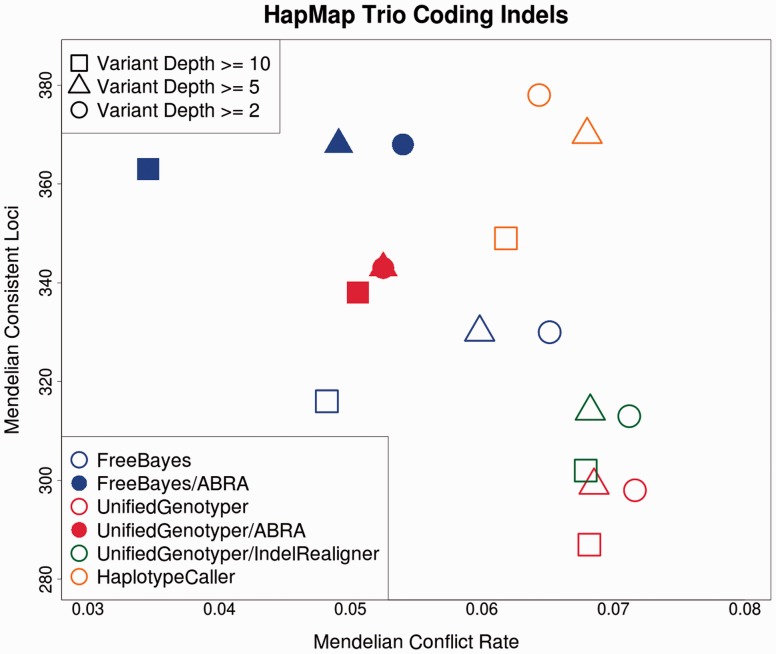
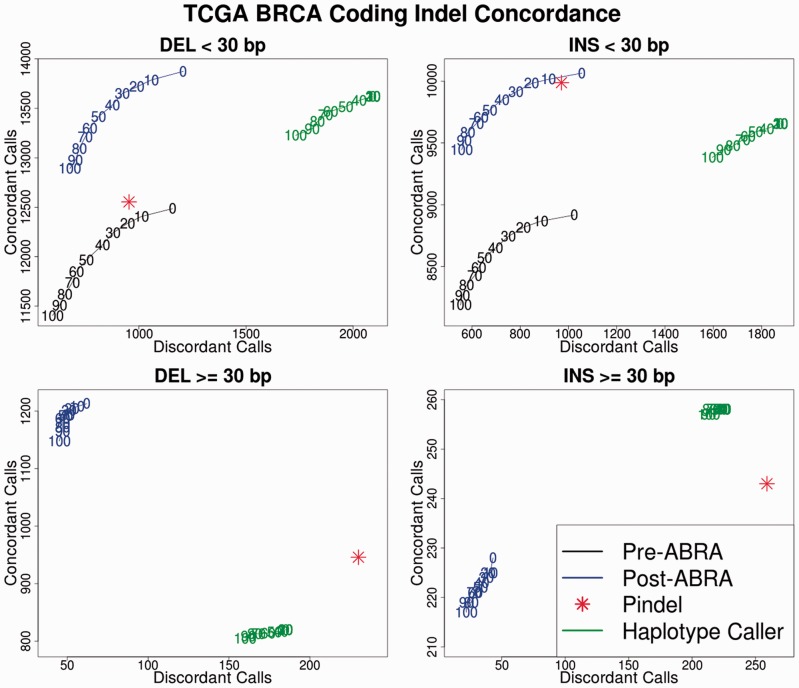
References
-
- Carnevali P, et al. Computational techniques for human genome resequencing using mated gapped reads. J. Comput. Biol. 2012;19:279–292. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
