

Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour
- PMID: 24909261
- PMCID: PMC4062040
- DOI: 10.1038/ncomms5039
Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour
Abstract
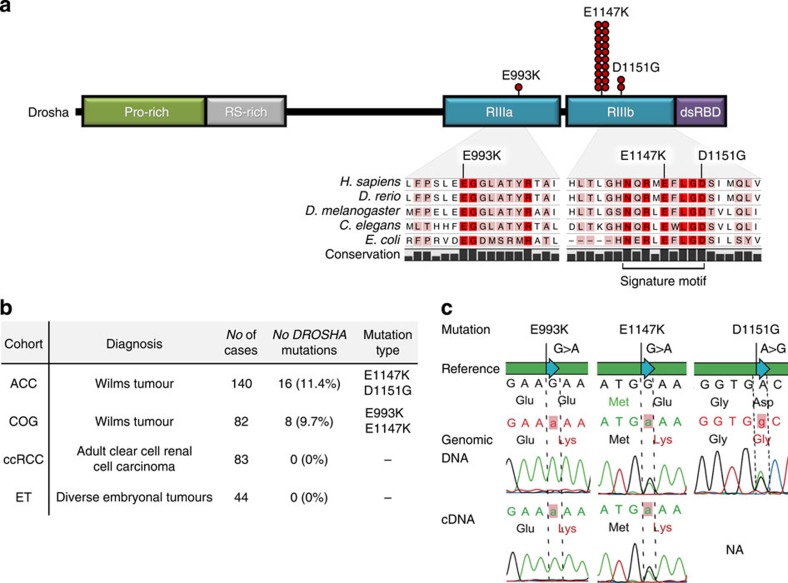
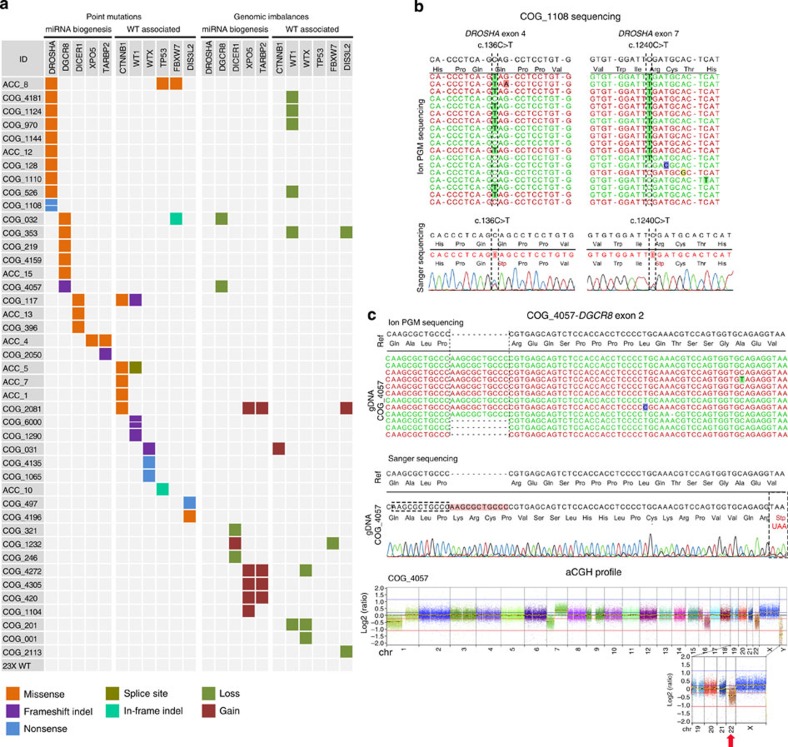
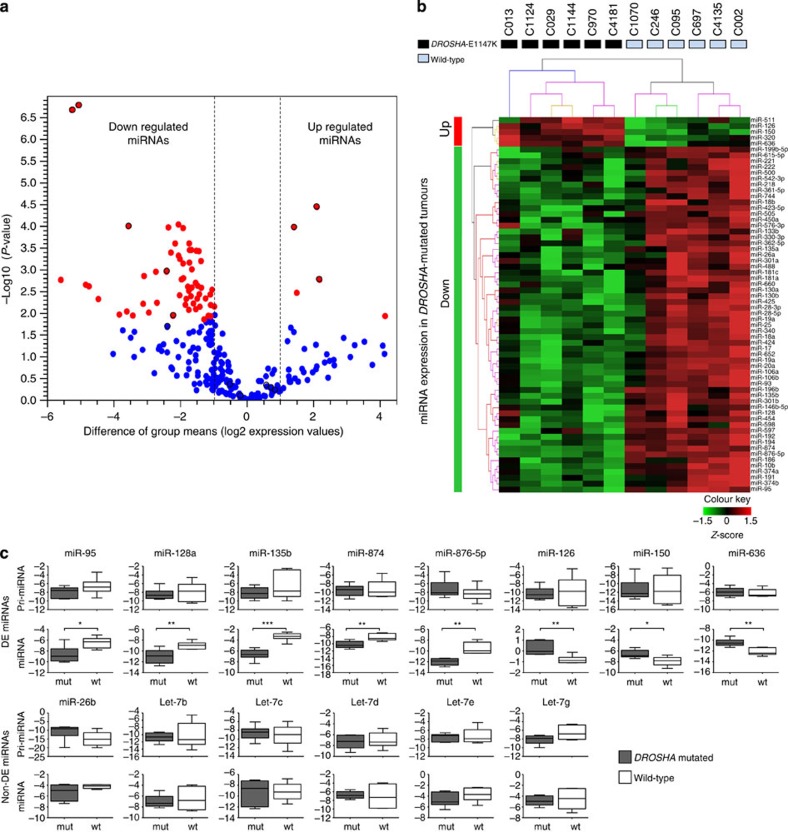
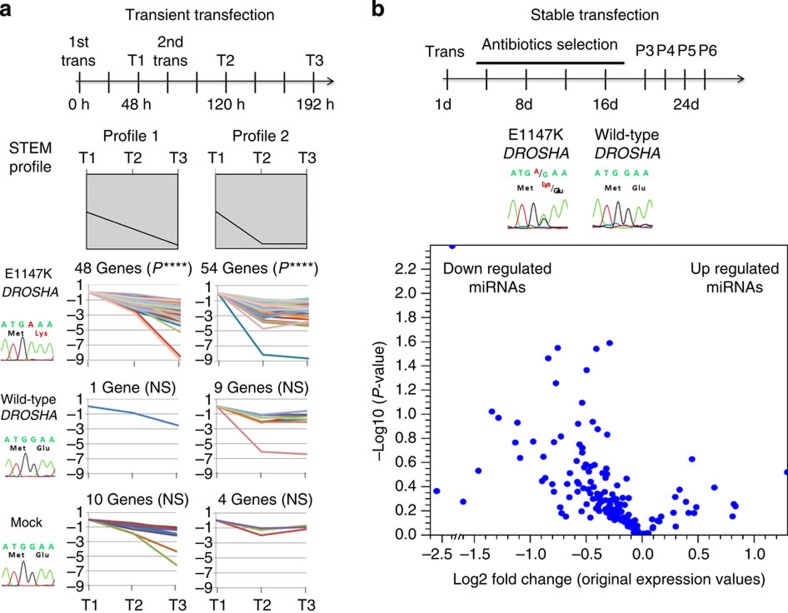
Wilms tumour (WT) is an embryonal kidney neoplasia for which very few driver genes have been identified. Here we identify DROSHA mutations in 12% of WT samples (26/222) using whole-exome sequencing and targeted sequencing of 10 microRNA (miRNA)-processing genes. A recurrent mutation (E1147K) affecting a metal-binding residue of the RNase IIIb domain is detected in 81% of the DROSHA-mutated tumours. In addition, we identify non-recurrent mutations in other genes of this pathway (DGCR8, DICER1, XPO5 and TARBP2). By assessing the miRNA expression pattern of the DROSHA-E1147K-mutated tumours and cell lines expressing this mutation, we determine that this variant leads to a predominant downregulation of a subset of miRNAs. We confirm that the downregulation occurs exclusively in mature miRNAs and not in primary miRNA transcripts, suggesting that the DROSHA E1147K mutation affects processing of primary miRNAs. Our data underscore the pivotal role of the miRNA biogenesis pathway in WT tumorigenesis, particularly the major miRNA-processing gene DROSHA.
Figures
References
-
- Honeyman J. N. et al. Factors associated with relapse and survival in Wilms tumor: a multivariate analysis. J. Pediatr. Surg. 47, 1228–1233 (2012). - PubMed
-
- Reinhard H. et al. Outcome of relapses of nephroblastoma in patients registered in the SIOP/GPOH trials and studies. Oncol. Rep. 20, 463–467 (2008). - PubMed
-
- Coppes M. J., Haber D. A. & Grundy P. E. Genetic events in the development of Wilms’ tumor. New Engl. J. Med. 331, 586–590 (1994). - PubMed
-
- Call K. M. et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 60, 509–520 (1990). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
