

Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions
- PMID: 24911462
- PMCID: PMC4346093
- DOI: 10.1016/j.jhep.2014.05.047
Vascular pathobiology in chronic liver disease and cirrhosis - current status and future directions
Abstract
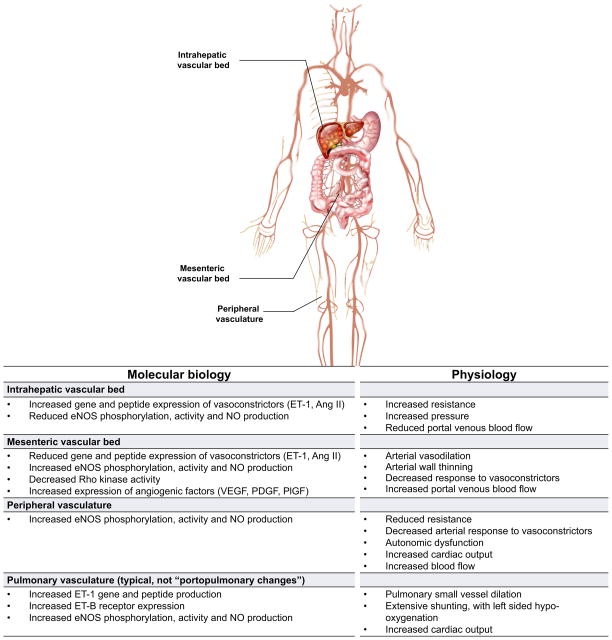
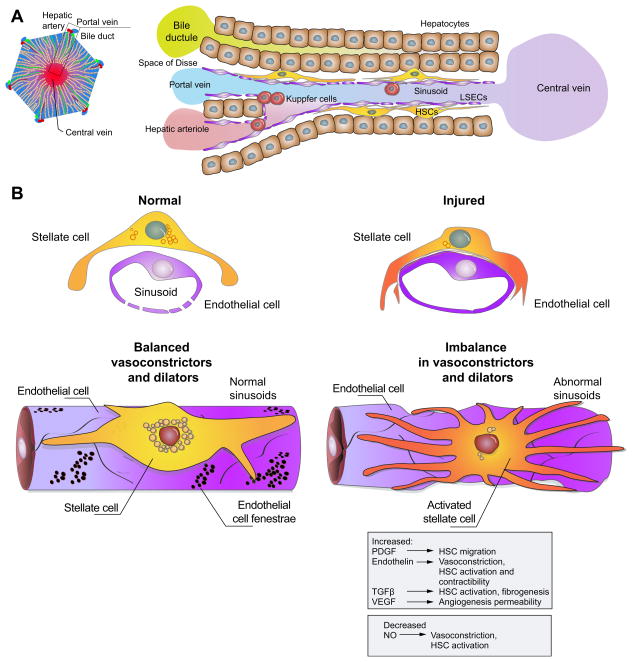
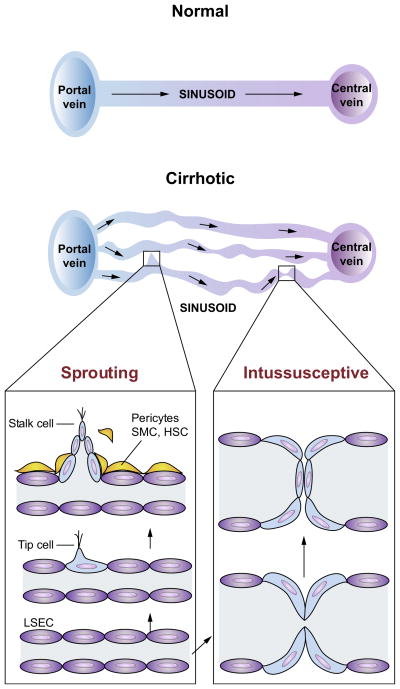
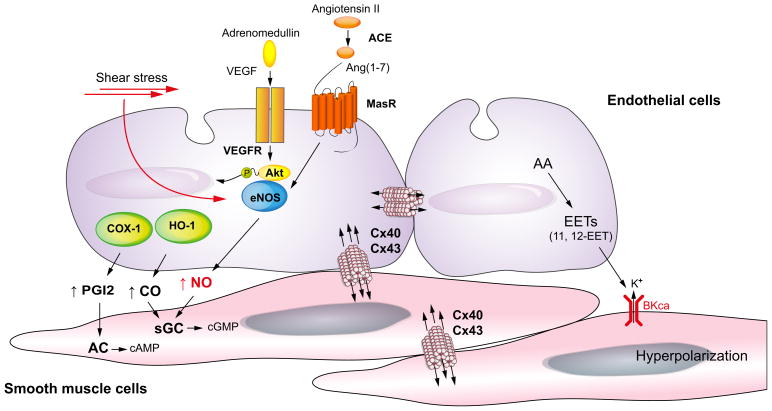
Chronic liver disease is associated with remarkable alterations in the intra- and extrahepatic vasculature. Because of these changes, the fields of liver vasculature and portal hypertension have recently become closely integrated within the broader vascular biology discipline. As developments in vascular biology have evolved, a deeper understanding of vascular processes has led to a better understanding of the mechanisms of the dynamic vascular changes associated with portal hypertension and chronic liver disease. In this context, hepatic vascular cells, such as sinusoidal endothelial cells and pericyte-like hepatic stellate cells, are closely associated with one another, where they have paracrine and autocrine effects on each other and themselves. These cells play important roles in the pathogenesis of liver fibrosis/cirrhosis and portal hypertension. Further, a variety of signaling pathways have recently come to light. These include growth factor pathways involving cytokines such as transforming growth factor β, platelet derived growth factor, and others as well as a variety of vasoactive peptides and other molecules. An early and consistent feature of liver injury is the development of an increase in intra-hepatic resistance; this is associated with changes in hepatic vascular cells and their signaling pathway that cause portal hypertension. A critical concept is that this process aggregates signals to the extrahepatic circulation, causing derangement in this system's cells and signaling pathways, which ultimately leads to the collateral vessel formation and arterial vasodilation in the splanchnic and systemic circulation, which by virtue of the hydraulic derivation of Ohm's law (pressure = resistance × flow), worsens portal hypertension. This review provides a detailed review of the current status and future direction of the basic biology of portal hypertension with a focus on the physiology, pathophysiology, and signaling of cells within the liver, as well as those in the mesenteric vascular circulation. Translational implications of recent research and the future directions that it points to are also highlighted.
Keywords: Endothelial cell; Pericyte; Pressure; Resistance; Sinusoid; Therapy.
Copyright © 2014 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Wisse E, De Zanger RB, Jacobs R, McCuskey RS. Scanning electron microscope observations on the structure of portal veins, sinusoids and central veins in rat liver. Scanning Microsc. 1983:1441–1452. - PubMed
-
- Bhunchet E, Fujieda K. Capillarization and venularization of hepatic sinusoids in porcine serum-induced rat liver fibrosis: a mechanism to maintain liver blood flow. Hepatology. 1993;18:1450–1458. - PubMed
-
- Shah V, Toruner M, Haddad F, Cadelina G, Papapetropoulos A, Choo K, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology. 1999;117:1222–1228. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
