

Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea
- PMID: 24916971
- PMCID: PMC4097860
- DOI: 10.1186/gb-2014-15-6-r77
Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea
Abstract
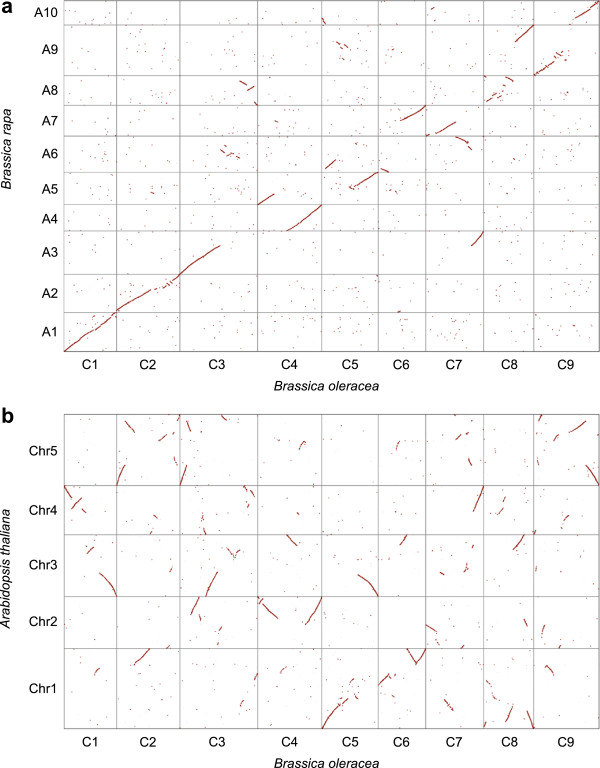
Background: Brassica oleracea is a valuable vegetable species that has contributed to human health and nutrition for hundreds of years and comprises multiple distinct cultivar groups with diverse morphological and phytochemical attributes. In addition to this phenotypic wealth, B. oleracea offers unique insights into polyploid evolution, as it results from multiple ancestral polyploidy events and a final Brassiceae-specific triplication event. Further, B. oleracea represents one of the diploid genomes that formed the economically important allopolyploid oilseed, Brassica napus. A deeper understanding of B. oleracea genome architecture provides a foundation for crop improvement strategies throughout the Brassica genus.
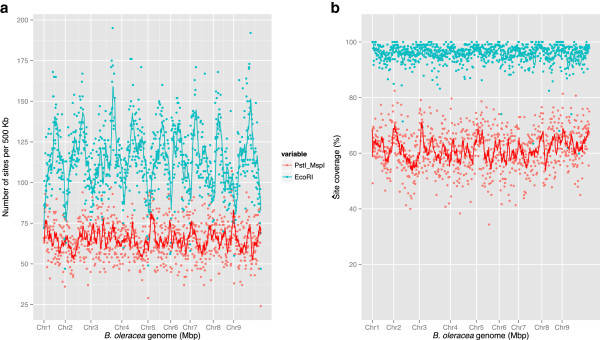
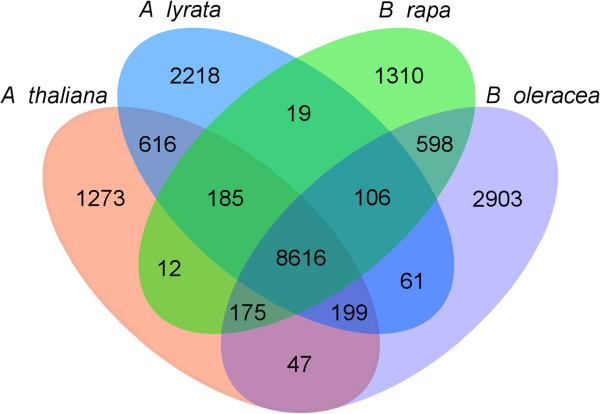
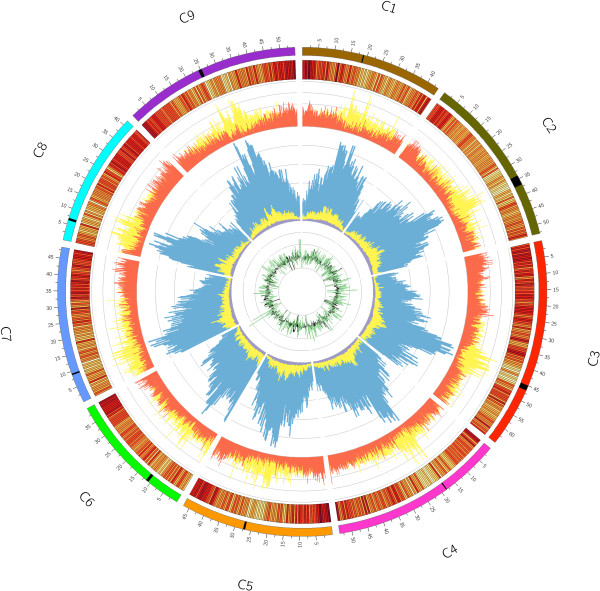
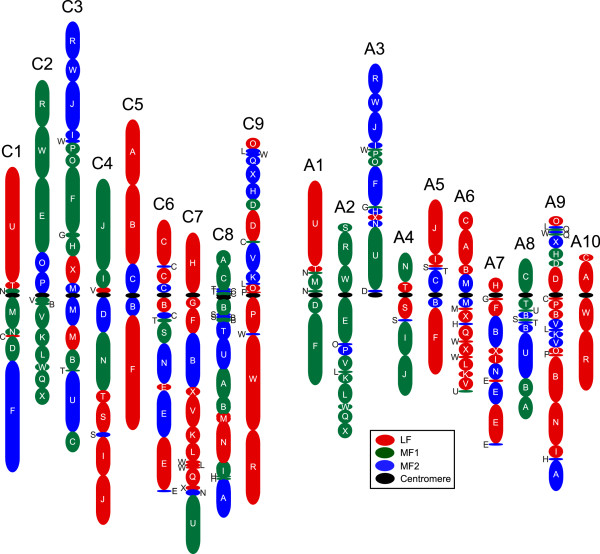
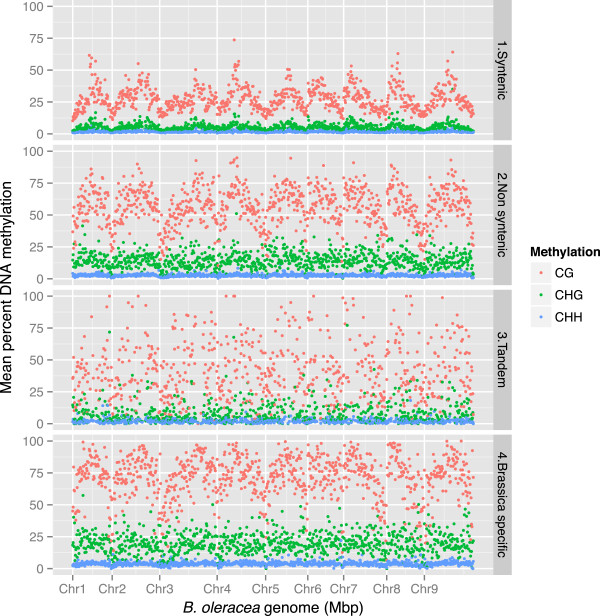
Results: We generate an assembly representing 75% of the predicted B. oleracea genome using a hybrid Illumina/Roche 454 approach. Two dense genetic maps are generated to anchor almost 92% of the assembled scaffolds to nine pseudo-chromosomes. Over 50,000 genes are annotated and 40% of the genome predicted to be repetitive, thus contributing to the increased genome size of B. oleracea compared to its close relative B. rapa. A snapshot of both the leaf transcriptome and methylome allows comparisons to be made across the triplicated sub-genomes, which resulted from the most recent Brassiceae-specific polyploidy event.
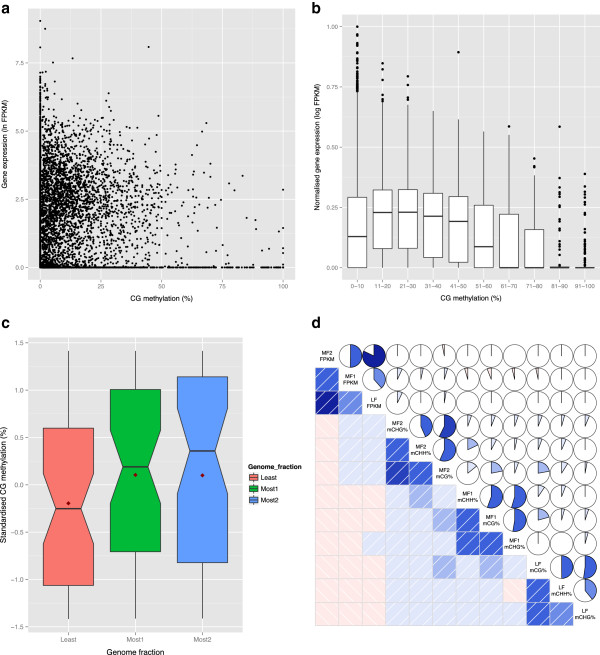
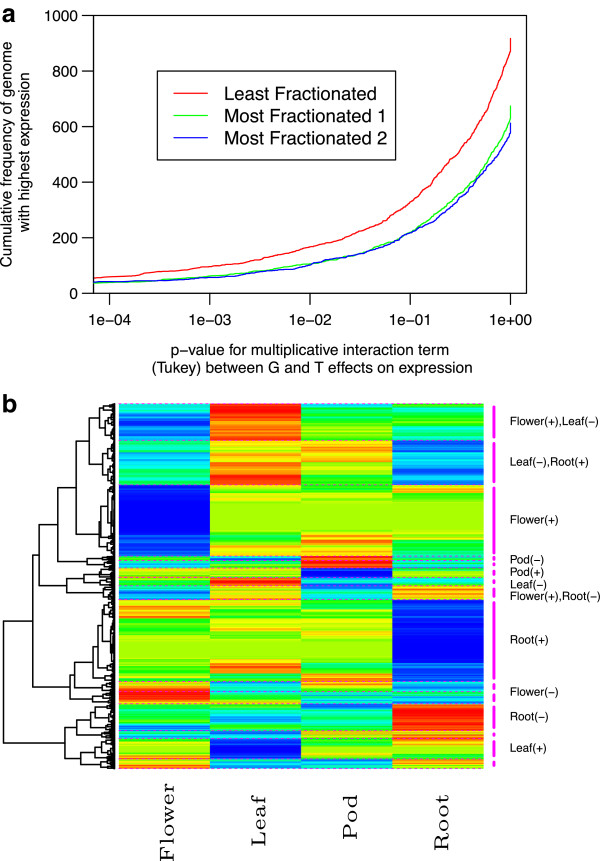
Conclusions: Differential expression of the triplicated syntelogs and cytosine methylation levels across the sub-genomes suggest residual marks of the genome dominance that led to the current genome architecture. Although cytosine methylation does not correlate with individual gene dominance, the independent methylation patterns of triplicated copies suggest epigenetic mechanisms play a role in the functional diversification of duplicate genes.
Figures
References
-
- Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun JH, Bancroft I, Cheng F, Huang S, Li X, Hua W, Wang J, Wang X, Freeling M, Pires JC, Paterson AH, Chalhoub B, Wang B, Hayward A, Sharpe AG, Park BS, Weisshaar B, Liu B, Li B, Liu B, Tong C, Song C, Duran C. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Gen. 2011;43:1035–1039. - PubMed
-
- Leitch AR, Leitch IJ. Genomic plasticity and the diversity of polyploid plants. Science. 2008;320:481–483. - PubMed
-
- Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422:433–438. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
