

Dysregulation of cellular iron metabolism in Friedreich ataxia: from primary iron-sulfur cluster deficit to mitochondrial iron accumulation
- PMID: 24917819
- PMCID: PMC4042101
- DOI: 10.3389/fphar.2014.00130
Dysregulation of cellular iron metabolism in Friedreich ataxia: from primary iron-sulfur cluster deficit to mitochondrial iron accumulation
Abstract
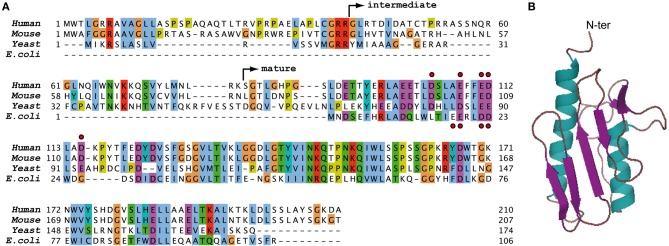
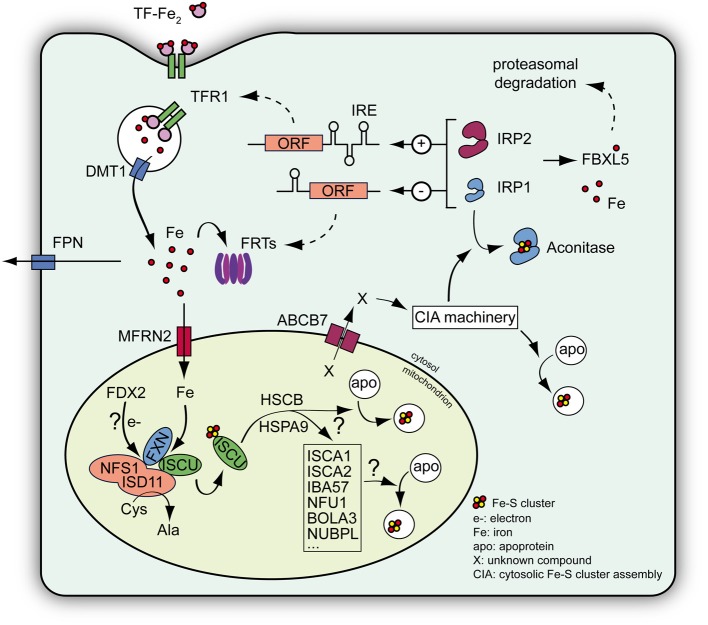
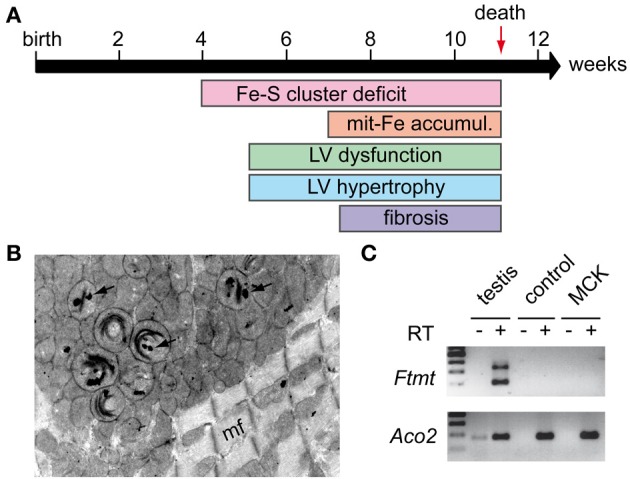
Friedreich ataxia (FRDA) is the most common recessive ataxia in the Caucasian population and is characterized by a mixed spinocerebellar and sensory ataxia frequently associating cardiomyopathy. The disease results from decreased expression of the FXN gene coding for the mitochondrial protein frataxin. Early histological and biochemical study of the pathophysiology in patient's samples revealed that dysregulation of iron metabolism is a key feature of the disease, mainly characterized by mitochondrial iron accumulation and by decreased activity of iron-sulfur cluster enzymes. In the recent past years, considerable progress in understanding the function of frataxin has been provided through cellular and biochemical approaches, pointing to the primary role of frataxin in iron-sulfur cluster biogenesis. However, why and how the impact of frataxin deficiency on this essential biosynthetic pathway leads to mitochondrial iron accumulation is still poorly understood. Herein, we review data on both the primary function of frataxin and the nature of the iron metabolism dysregulation in FRDA. To date, the pathophysiological implication of the mitochondrial iron overload in FRDA remains to be clarified.
Keywords: Friedreich ataxia; frataxin; iron metabolism; iron metabolism disorders; iron-sulfur cluster; mitochondria.
Figures
References
-
- Al-Mahdawi S., Pinto R. M., Varshney D., Lawrence L., Lowrie M. B., Hughes S., et al. (2006). GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 88, 580–590 10.1016/j.ygeno.2006.06.015 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous