

Strain- and plasmid-level deconvolution of a synthetic metagenome by sequencing proximity ligation products
- PMID: 24918035
- PMCID: PMC4045339
- DOI: 10.7717/peerj.415
Strain- and plasmid-level deconvolution of a synthetic metagenome by sequencing proximity ligation products
Abstract
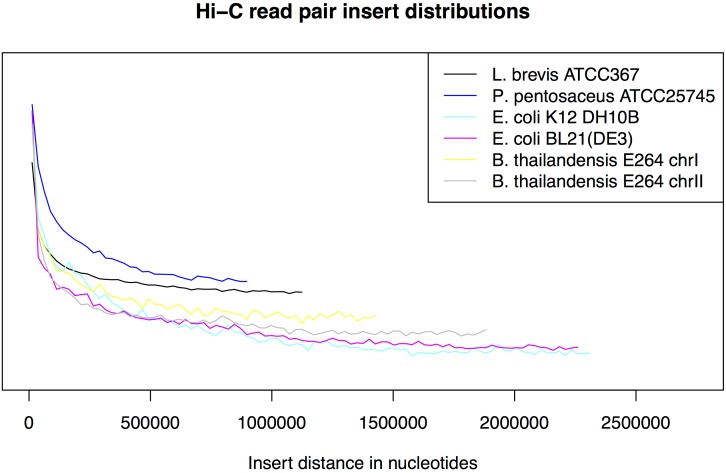
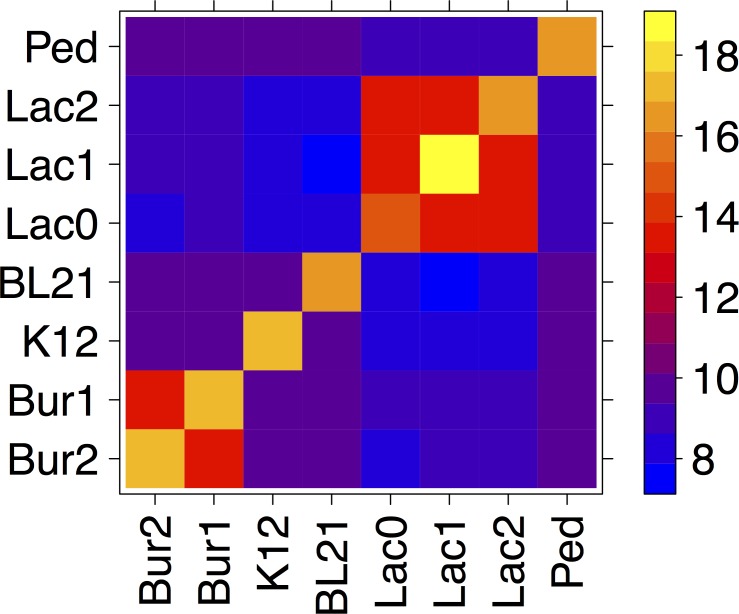
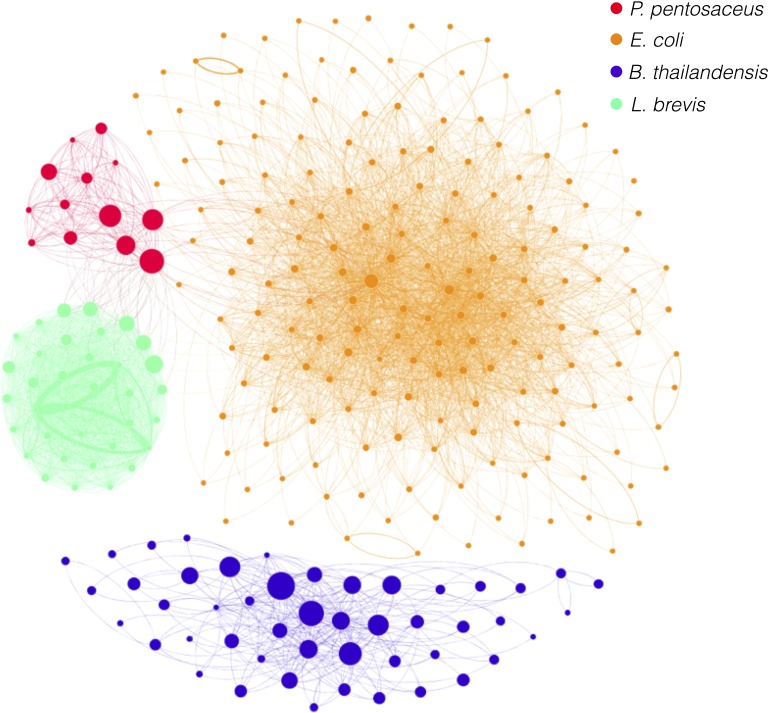
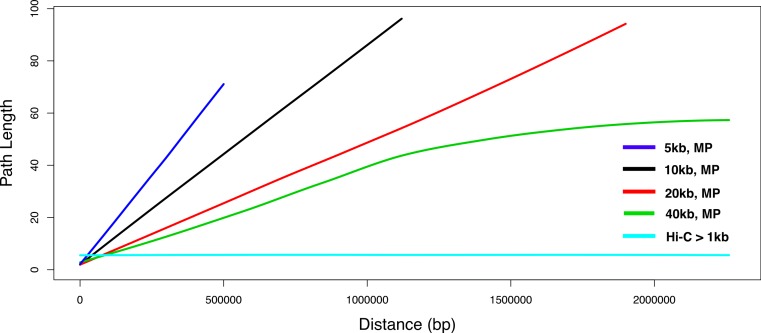
Metagenomics is a valuable tool for the study of microbial communities but has been limited by the difficulty of "binning" the resulting sequences into groups corresponding to the individual species and strains that constitute the community. Moreover, there are presently no methods to track the flow of mobile DNA elements such as plasmids through communities or to determine which of these are co-localized within the same cell. We address these limitations by applying Hi-C, a technology originally designed for the study of three-dimensional genome structure in eukaryotes, to measure the cellular co-localization of DNA sequences. We leveraged Hi-C data generated from a simple synthetic metagenome sample to accurately cluster metagenome assembly contigs into groups that contain nearly complete genomes of each species. The Hi-C data also reliably associated plasmids with the chromosomes of their host and with each other. We further demonstrated that Hi-C data provides a long-range signal of strain-specific genotypes, indicating such data may be useful for high-resolution genotyping of microbial populations. Our work demonstrates that Hi-C sequencing data provide valuable information for metagenome analyses that are not currently obtainable by other methods. This metagenomic Hi-C method could facilitate future studies of the fine-scale population structure of microbes, as well as studies of how antibiotic resistance plasmids (or other genetic elements) mobilize in microbial communities. The method is not limited to microbiology; the genetic architecture of other heterogeneous populations of cells could also be studied with this technique.
Keywords: Genome scaffolding; Haplotype phasing; Hi-C; Markov clustering; Metagenome assembly; Metagenomics; Microbial ecology; Plasmids; Strain differentiation; Synthetic microbial communities.
Figures

References
-
- Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks. In: Adar E, Hurst M, Finin T, Glance NS, Nicolov N, Tseng BL, editors. The international conference on weblogs and social media. The AAAI Press; 2009.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
