

Mutation G805R in the transmembrane domain of the LDL receptor gene causes familial hypercholesterolemia by inducing ectodomain cleavage of the LDL receptor in the endoplasmic reticulum
- PMID: 24918045
- PMCID: PMC4048843
- DOI: 10.1016/j.fob.2014.03.007
Mutation G805R in the transmembrane domain of the LDL receptor gene causes familial hypercholesterolemia by inducing ectodomain cleavage of the LDL receptor in the endoplasmic reticulum
Abstract
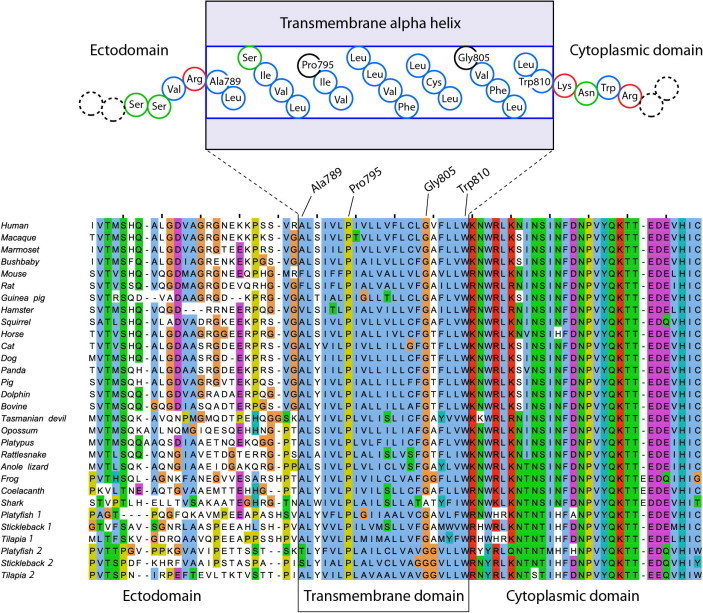
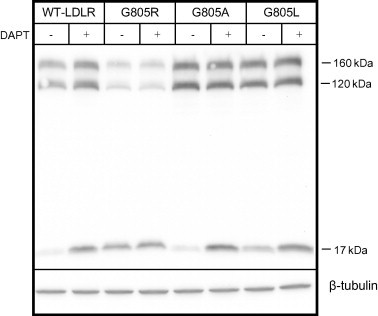
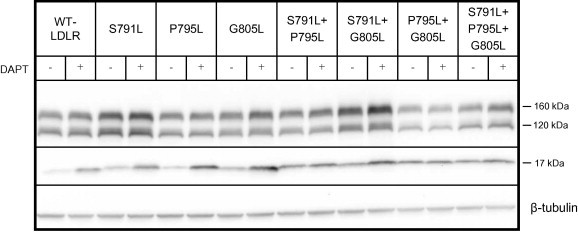
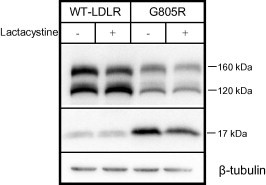
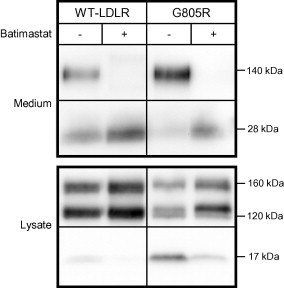
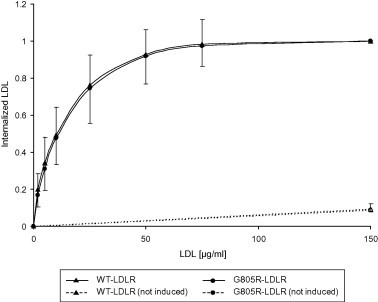
More than 1700 mutations in the low density lipoprotein receptor (LDLR) gene have been found to cause familial hypercholesterolemia (FH). These are commonly divided into five classes based upon their effects on the structure and function of the LDLR. However, little is known about the mechanism by which mutations in the transmembrane domain of the LDLR gene cause FH. We have studied how the transmembrane mutation G805R affects the function of the LDLR. Based upon Western blot analyses of transfected HepG2 cells, mutation G805R reduced the amounts of the 120 kDa precursor LDLR in the endoplasmic reticulum. This led to reduced amounts of the mature 160 kDa LDLR at the cell surface. However, significant amounts of a secreted 140 kDa G805R-LDLR ectodomain fragment was observed in the culture media. Treatment of the cells with the metalloproteinase inhibitor batimastat largely restored the amounts of the 120 and 160 kDa forms in cell lysates, and prevented secretion of the 140 kDa ectodomain fragment. Together, these data indicate that a metalloproteinase cleaved the ectodomain of the 120 kDa precursor G805R-LDLR in the endoplasmic reticulum. It was the presence of the polar Arg805 and not the lack of Gly805 which led to ectodomain cleavage. Arg805 also prevented γ-secretase cleavage within the transmembrane domain. It is conceivable that introducing a charged residue within the hydrophobic membrane lipid bilayer, results in less efficient incorporation of the 120 kDa G805R-LDLR in the endoplasmic reticulum membrane and makes it a substrate for metalloproteinase cleavage.
Keywords: DAPT, N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester; DiD, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate; Endoplasmic reticulum; Familial hypercholesterolemia; LDL receptor; LDL, low density lipoprotein; LDLR, low density lipoprotein receptor; Metalloproteinase; Mutation; Transmembrane domain.
Figures

References
-
- Goldstein J.L., Hobbs H.H., Brown M.S. Familial hypercholesterolemia. In: Scriver C., Beaudet A.L., Sly W.S., Valle D., editors. The metabolic & molecular basis of inherited disease. McGraw-Hill; New York: 2001. pp. 2863–2914.
-
- Tolleshaug H., Goldstein J.L., Schneider W.J., Brown M.S. Posttranslational processing of the LDL receptor and its genetic disruption in familial hypercholesterolemia. Cell. 1982;30:715–724. - PubMed
-
- Brown M.S., Goldstein J.L. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. - PubMed
-
- Hobbs H.H., Russell D.W., Brown M.S., Goldstein J.L. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu. Rev. Genet. 1990;24:133–170. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
