

Single-cell RNA-seq reveals dynamic paracrine control of cellular variation
- PMID: 24919153
- PMCID: PMC4193940
- DOI: 10.1038/nature13437
Single-cell RNA-seq reveals dynamic paracrine control of cellular variation
Abstract
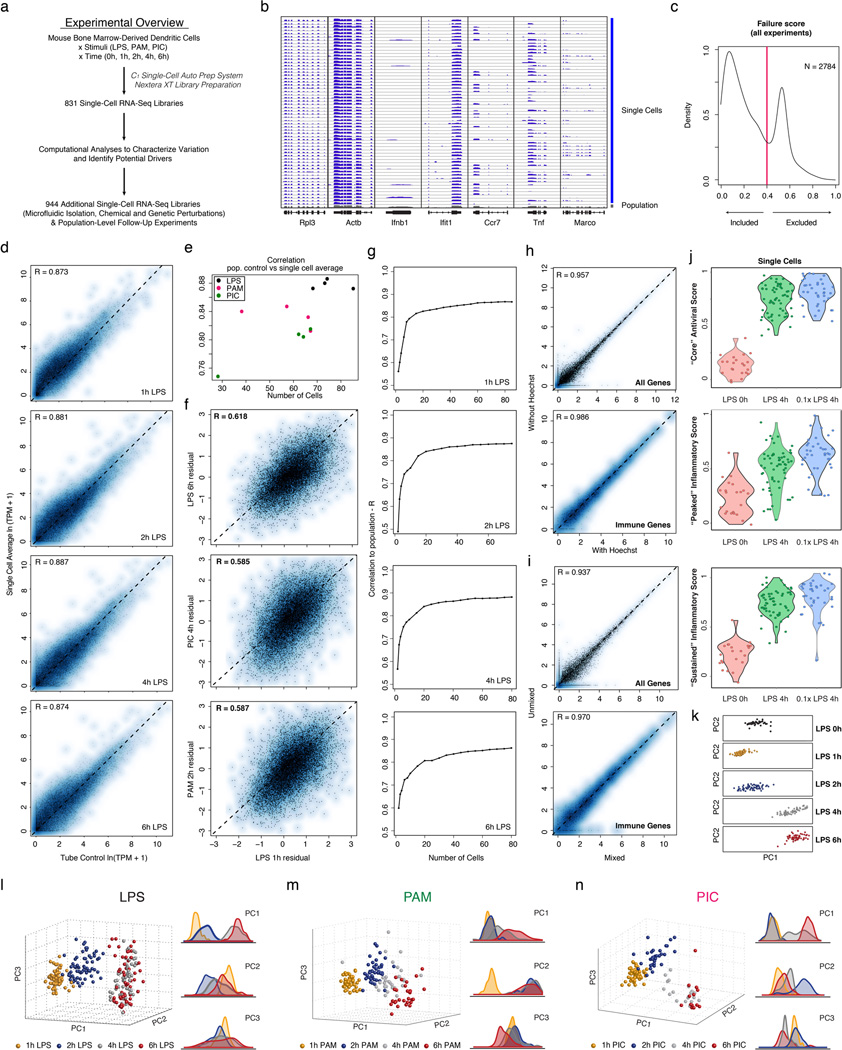
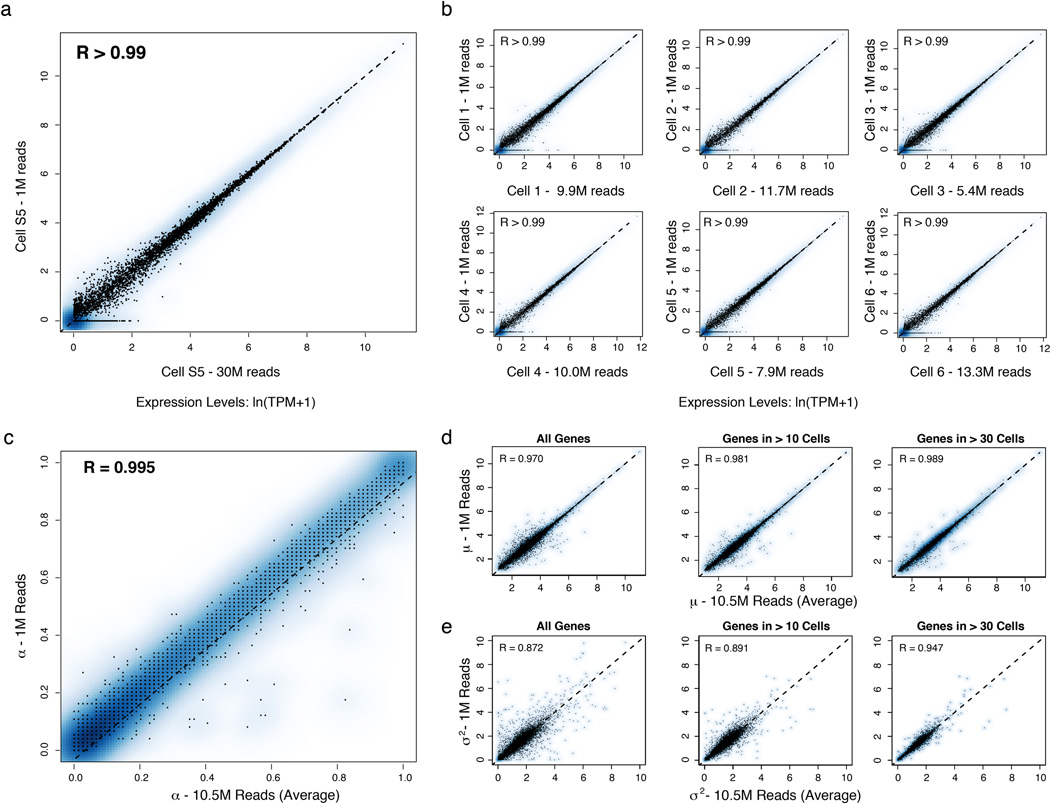
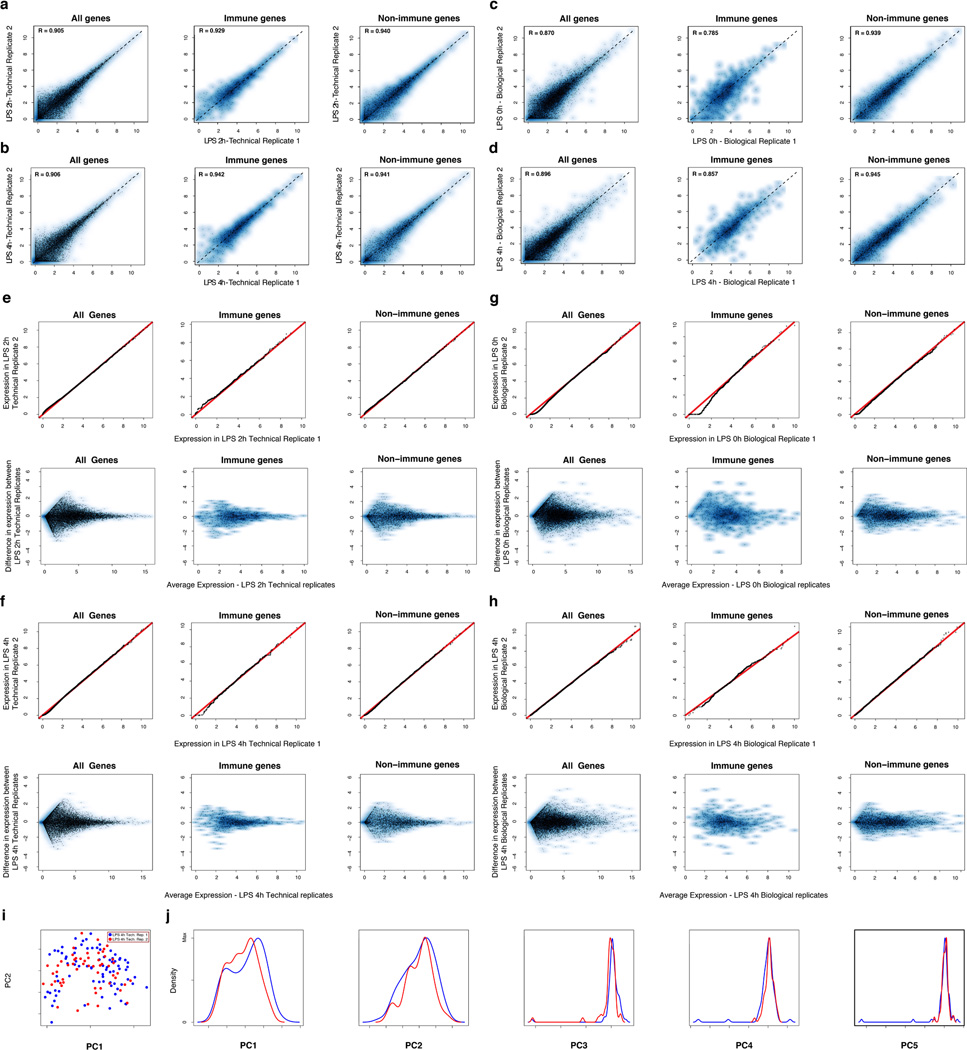
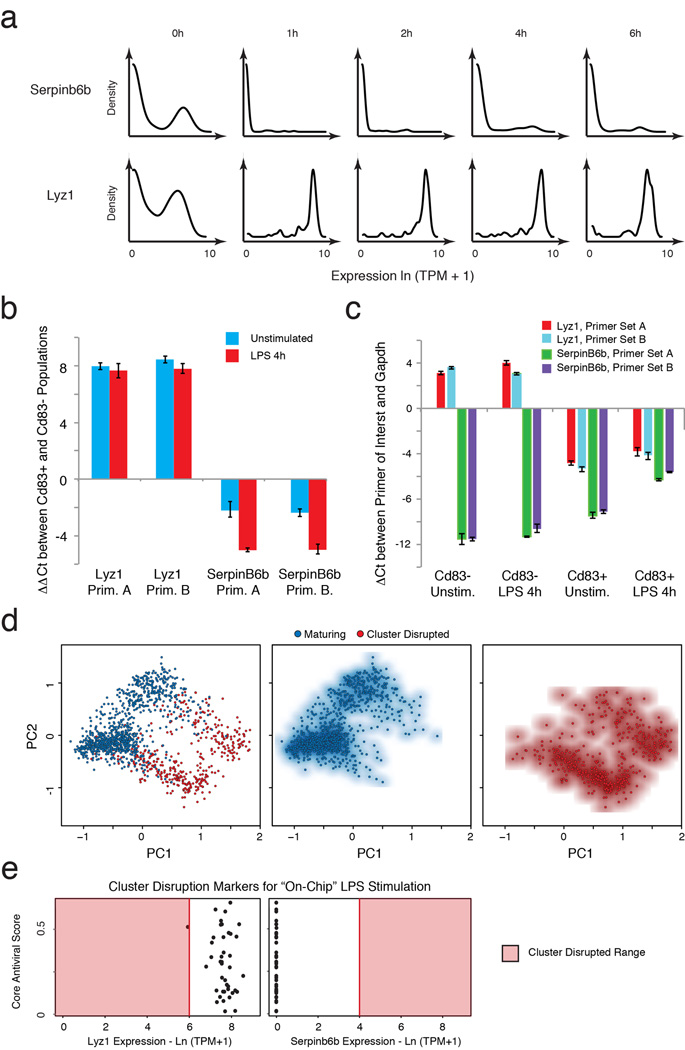
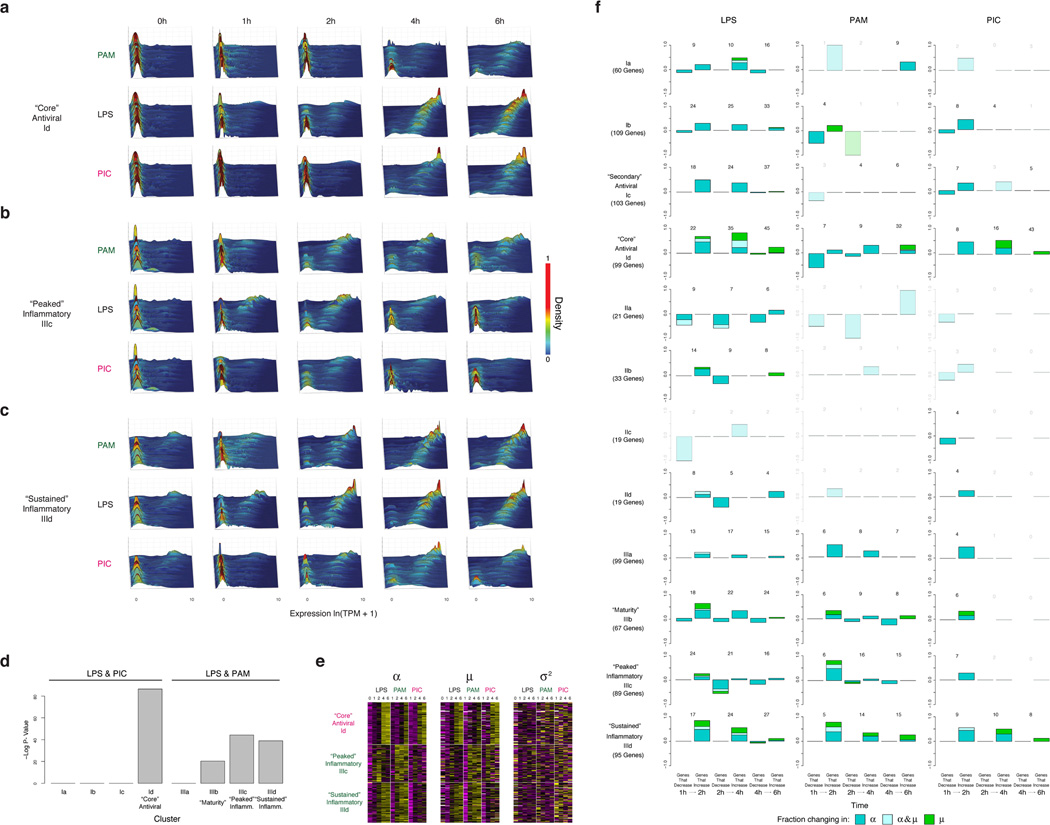
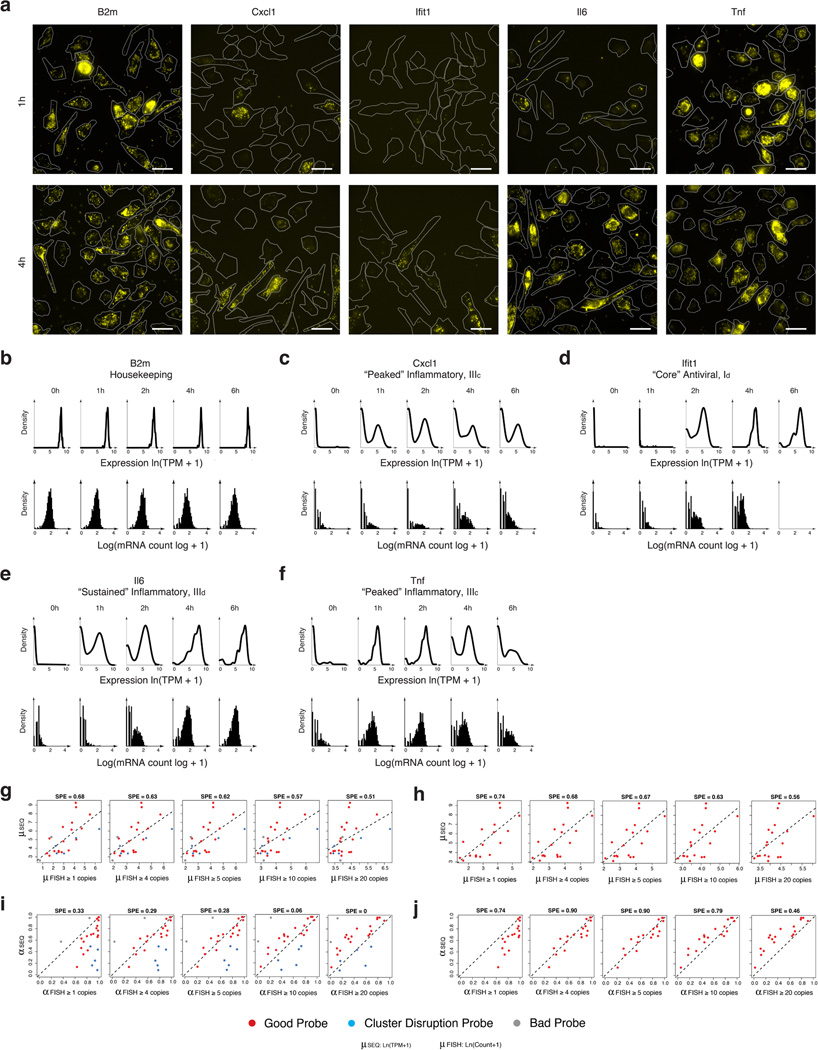
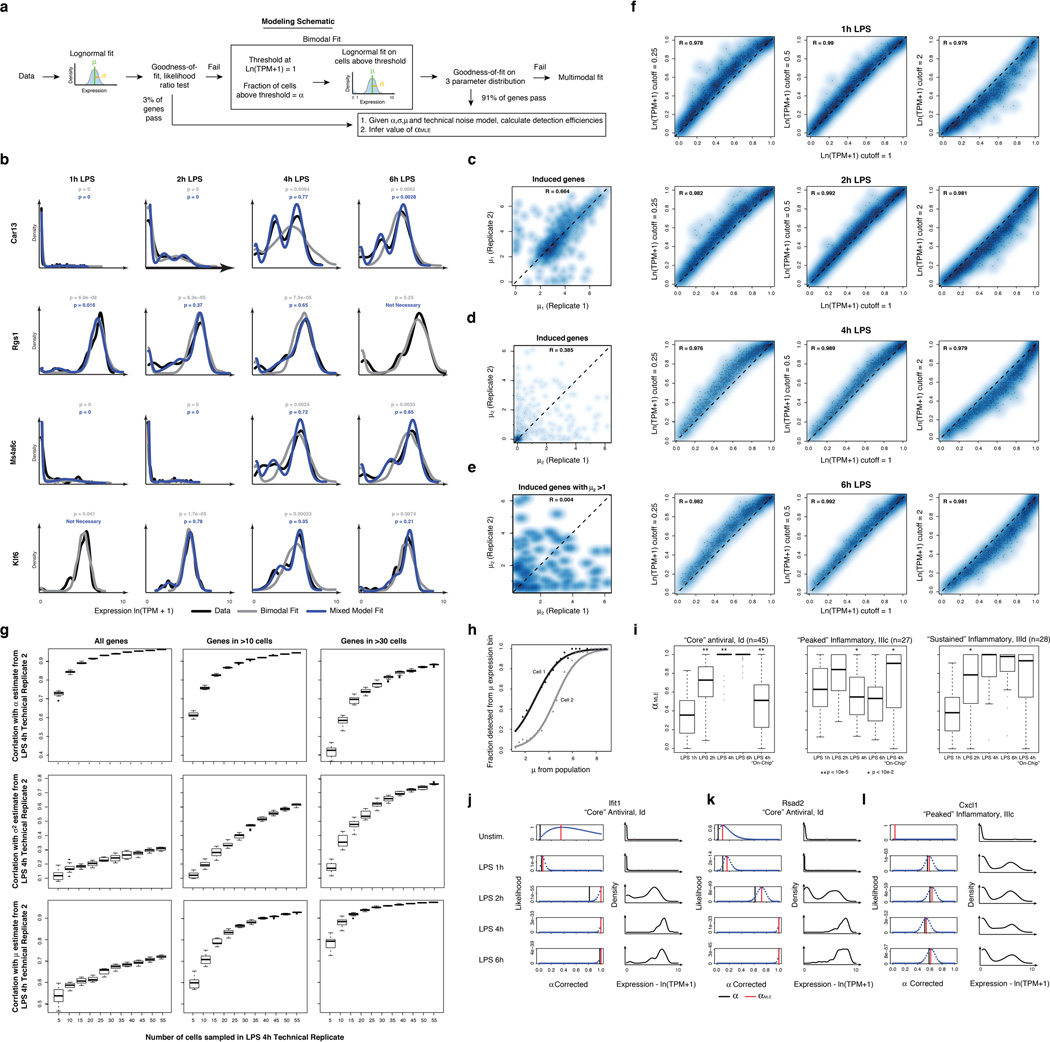
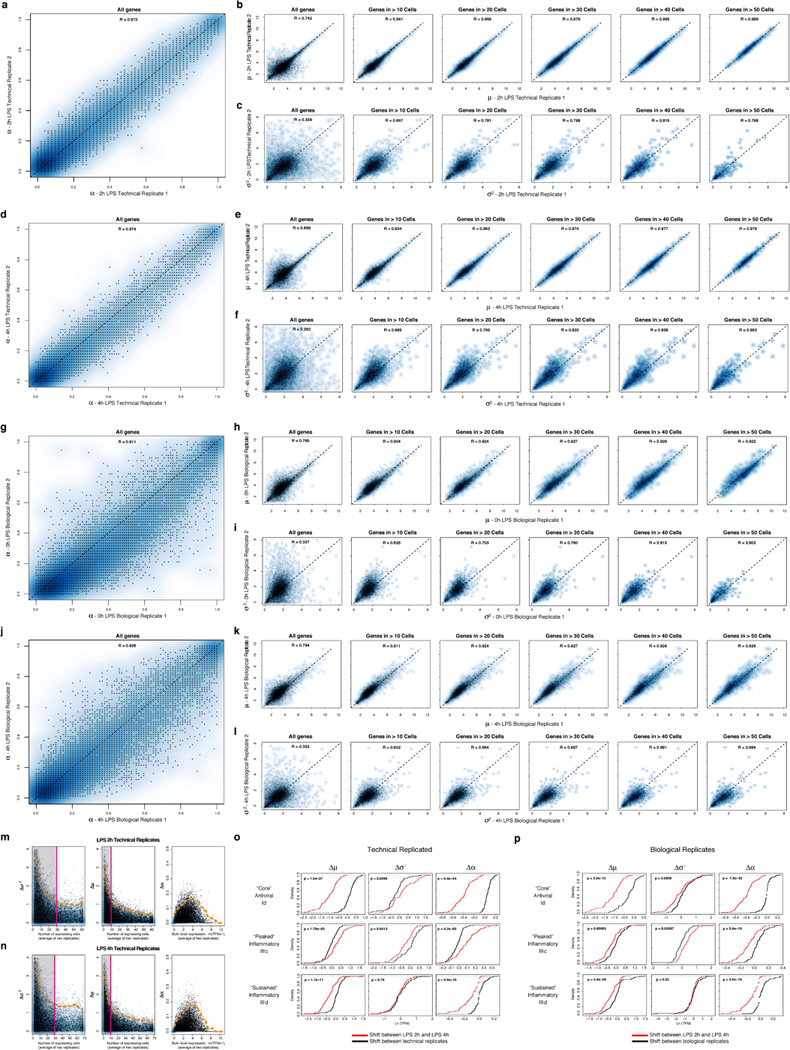
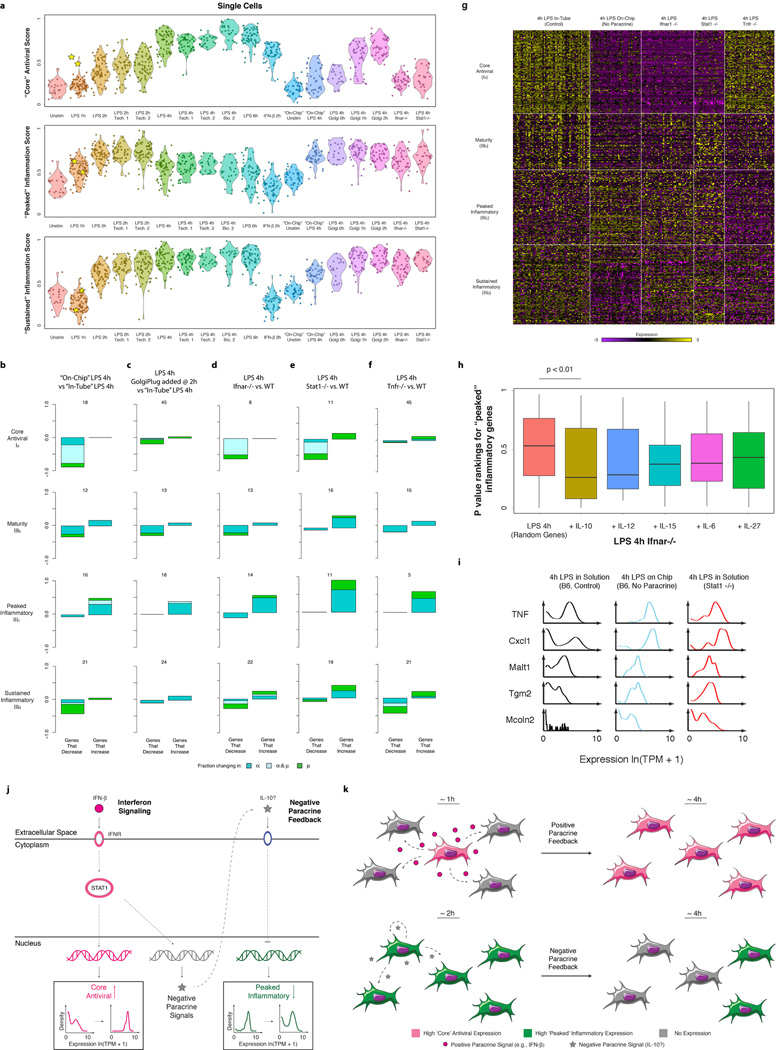
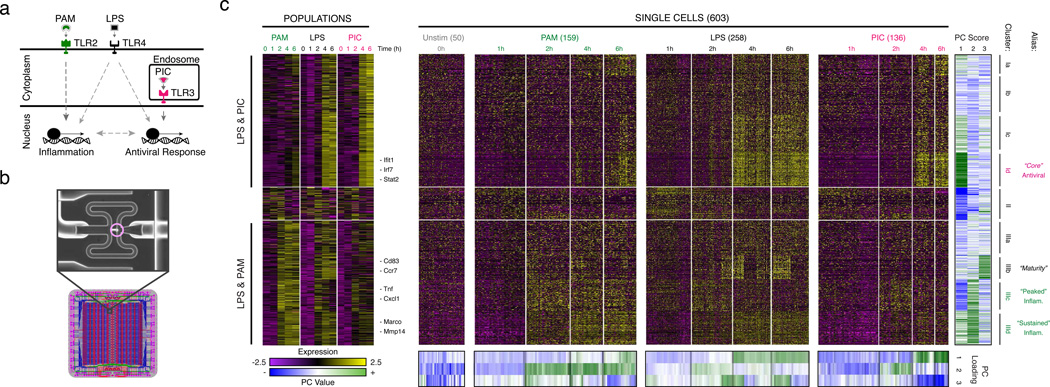
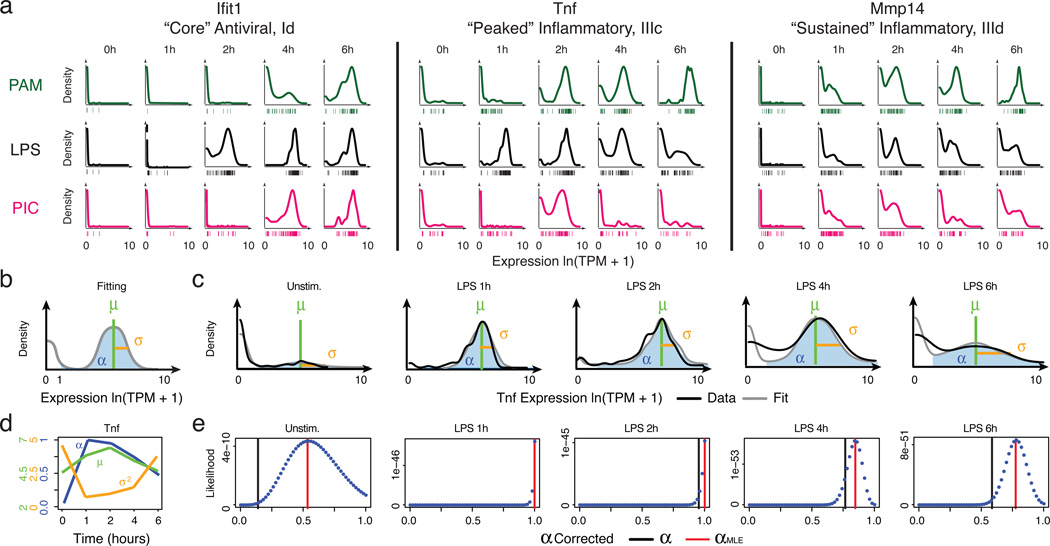
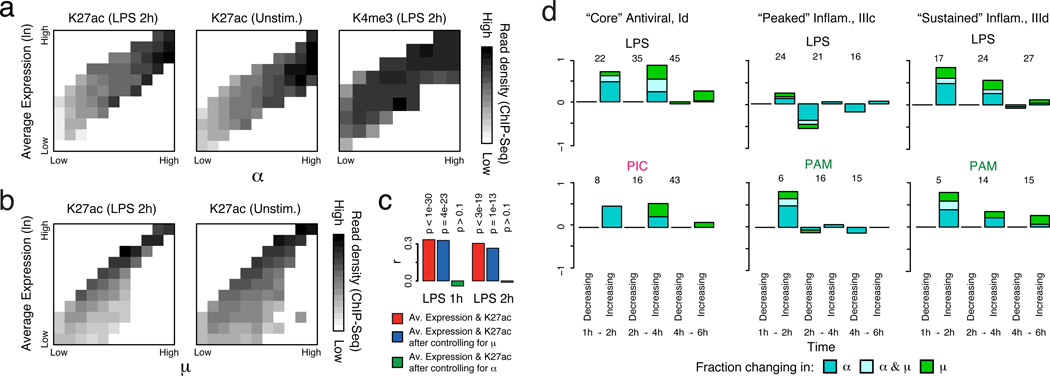
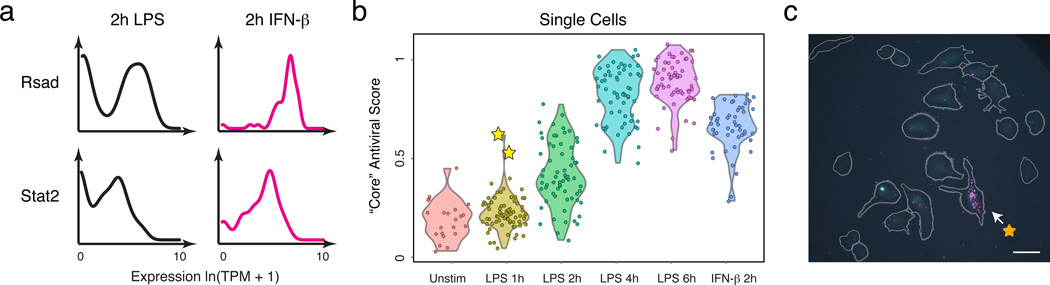
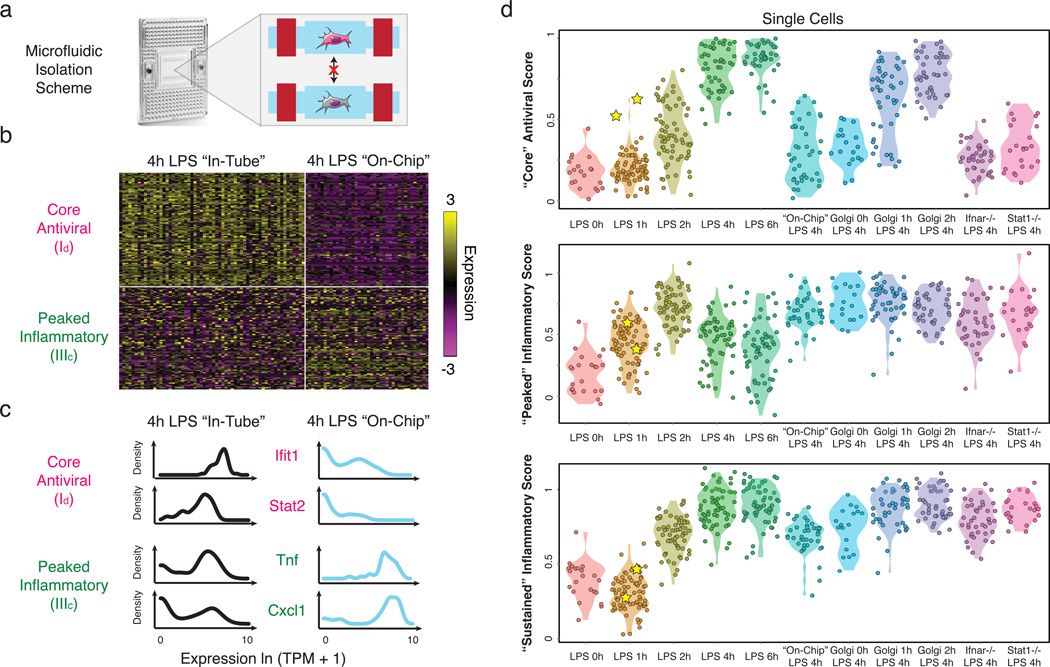
High-throughput single-cell transcriptomics offers an unbiased approach for understanding the extent, basis and function of gene expression variation between seemingly identical cells. Here we sequence single-cell RNA-seq libraries prepared from over 1,700 primary mouse bone-marrow-derived dendritic cells spanning several experimental conditions. We find substantial variation between identically stimulated dendritic cells, in both the fraction of cells detectably expressing a given messenger RNA and the transcript's level within expressing cells. Distinct gene modules are characterized by different temporal heterogeneity profiles. In particular, a 'core' module of antiviral genes is expressed very early by a few 'precocious' cells in response to uniform stimulation with a pathogenic component, but is later activated in all cells. By stimulating cells individually in sealed microfluidic chambers, analysing dendritic cells from knockout mice, and modulating secretion and extracellular signalling, we show that this response is coordinated by interferon-mediated paracrine signalling from these precocious cells. Notably, preventing cell-to-cell communication also substantially reduces variability between cells in the expression of an early-induced 'peaked' inflammatory module, suggesting that paracrine signalling additionally represses part of the inflammatory program. Our study highlights the importance of cell-to-cell communication in controlling cellular heterogeneity and reveals general strategies that multicellular populations can use to establish complex dynamic responses.
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
