

Genetic deficiency of neuronal RAGE protects against AGE-induced synaptic injury
- PMID: 24922072
- PMCID: PMC4611721
- DOI: 10.1038/cddis.2014.248
Genetic deficiency of neuronal RAGE protects against AGE-induced synaptic injury
Abstract
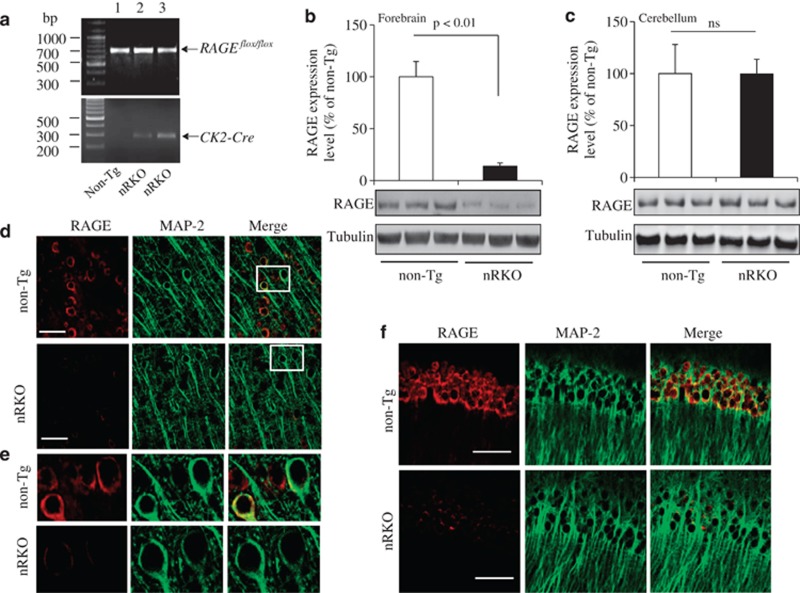
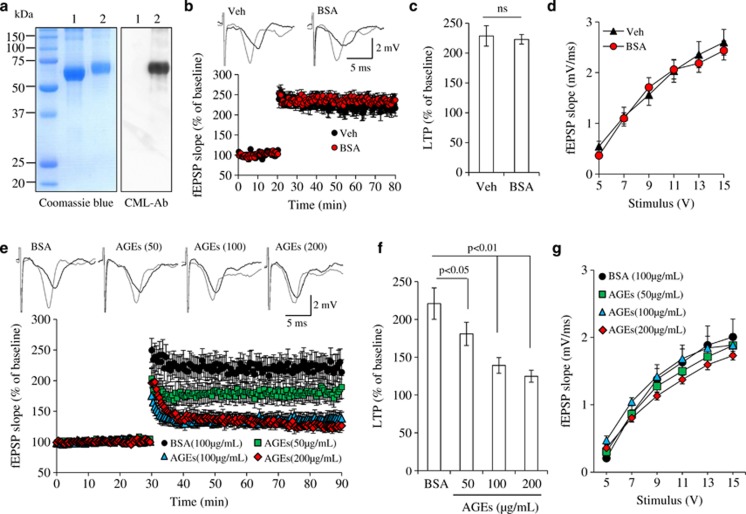
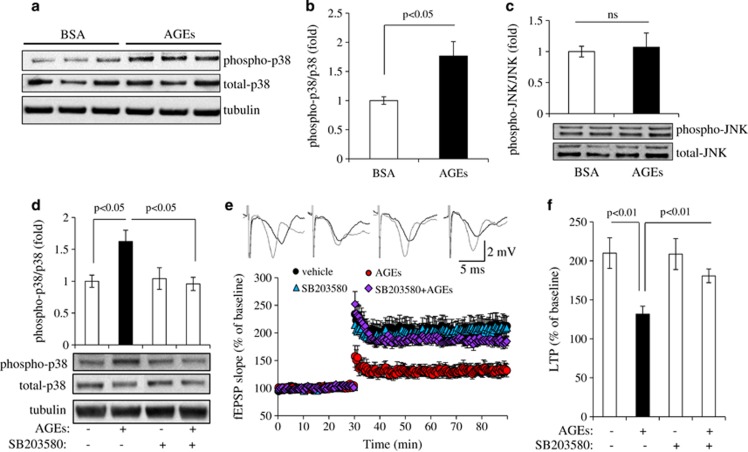
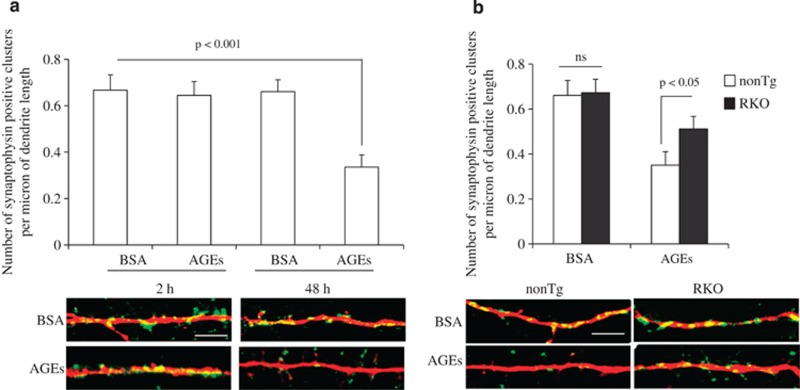
Synaptic dysfunction and degeneration is an early pathological feature of aging and age-related diseases, including Alzheimer's disease (AD). Aging is associated with increased generation and deposition of advanced glycation endproducts (AGEs), resulting from nonenzymatic glycation (or oxidation) proteins and lipids. AGE formation is accelerated in diabetes and AD-affected brain, contributing to cellular perturbation. The extent of AGEs' involvement, if at all, in alterations in synaptic structure and function is currently unknown. Here we analyze the contribution of neuronal receptor of AGEs (RAGE) signaling to AGE-mediated synaptic injury using novel transgenic neuronal RAGE knockout mice specifically targeted to the forebrain and transgenic mice expressing neuronal dominant-negative RAGE (DN-RAGE). Addition of AGEs to brain slices impaired hippocampal long-term potentiation (LTP). Similarly, treatment of hippocampal neurons with AGEs significantly decreases synaptic density. Such detrimental effects are largely reversed by genetic RAGE depletion. Notably, brain slices from mice with neuronal RAGE deficiency or DN-RAGE are resistant to AGE-induced LTP deficit. Further, RAGE deficiency or DN-RAGE blocks AGE-induced activation of p38 signaling. Taken together, these data show that neuronal RAGE functions as a signal transducer for AGE-induced synaptic dysfunction, thereby providing new insights into a mechanism by which the AGEs-RAGE-dependent signaling cascade contributes to synaptic injury via the p38 MAP kinase signal transduction pathway. Thus, RAGE blockade may be a target for development of interventions aimed at preventing the progression of cognitive decline in aging and age-related neurodegenerative diseases.
Figures



References
-
- 1Li J, Liu D, Sun L, Lu Y, Zhang Z. Advanced glycation end products and neurodegenerative diseases: mechanisms and perspective. J Neurol Sci 2012; 317: 1–5. - PubMed
-
- 2Munch G, Thome J, Foley P, Schinzel R, Riederer P. Advanced glycation endproducts in ageing and Alzheimer's disease. Brain Res Brain Res Rev 1997; 23: 134–143. - PubMed
-
- 3Vlassara H, Striker GE. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinol Metab Clin North Am 2013; 42: 697–719. - PubMed
-
- 4Takedo A, Yasuda T, Miyata T, Mizuno K, Li M, Yoneyama S et al. Immunohistochemical study of advanced glycation end products in aging and Alzheimer's disease brain. Neurosci Lett 1996; 221: 17–20. - PubMed
-
- 5Luth HJ, Ogunlade V, Kuhla B, Kientsch-Engel R, Stahl P, Webster J et al. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer's disease brains. Cereb Cortex 2005; 15: 211–220. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
