

Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis
- PMID: 24922578
- PMCID: PMC4071223
- DOI: 10.1016/j.chom.2014.05.012
Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis
Abstract
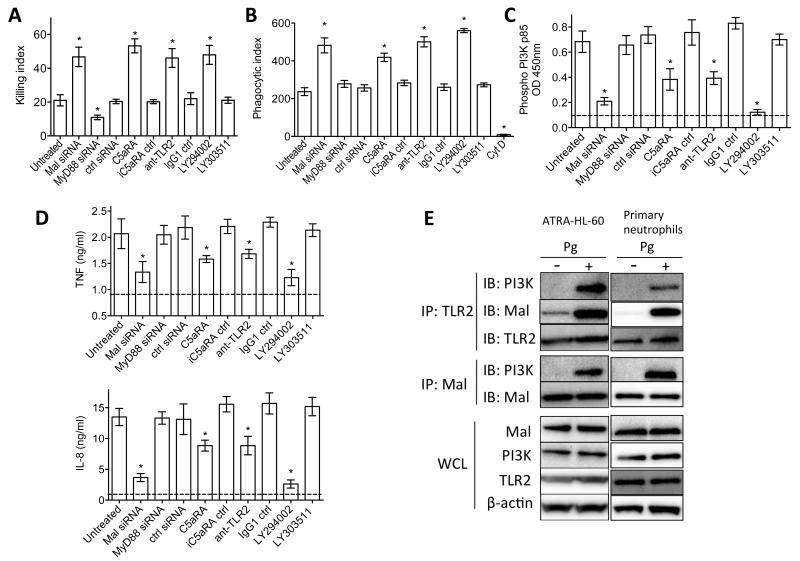
Certain low-abundance bacterial species, such as the periodontitis-associated oral bacterium Porphyromonas gingivalis, can subvert host immunity to remodel a normally symbiotic microbiota into a dysbiotic, disease-provoking state. However, such pathogens also exploit inflammation to thrive in dysbiotic conditions. How these bacteria evade immunity while maintaining inflammation is unclear. As previously reported, P. gingivalis remodels the oral microbiota into a dysbiotic state by exploiting complement. Now we show that in neutrophils P. gingivalis disarms a host-protective TLR2-MyD88 pathway via proteasomal degradation of MyD88, whereas it activates an alternate TLR2-Mal-PI3K pathway. This alternate TLR2-Mal-PI3K pathway blocks phagocytosis, provides "bystander" protection to otherwise susceptible bacteria, and promotes dysbiotic inflammation in vivo. This mechanism to disengage bacterial clearance from inflammation required an intimate crosstalk between TLR2 and the complement receptor C5aR and can contribute to the persistence of microbial communities that drive dysbiotic diseases.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures






Comment in
-
Porphyromonas gingivalis neutrophil manipulation: risk factor for periodontitis?Trends Microbiol. 2014 Aug;22(8):428-9. doi: 10.1016/j.tim.2014.06.006. Epub 2014 Jul 4. Trends Microbiol. 2014. PMID: 25001854 Free PMC article.
-
Mucosal immunology: two routes to success.Nat Rev Immunol. 2014 Aug;14(8):519. doi: 10.1038/nri3723. Epub 2014 Jul 18. Nat Rev Immunol. 2014. PMID: 25033908 No abstract available.
References
-
- Brzezinska AA, Johnson JL, Munafo DB, Ellis BA, Catz SD. Signalling mechanisms for Toll-like receptor-activated neutrophil exocytosis: key roles for interleukin-1-receptor-associated kinase-4 and phosphatidylinositol 3-kinase but not Toll/IL-1 receptor (TIR) domain-containing adaptor inducing IFNβ (TRIF) Immunology. 2009;127:386–397. - PMC - PubMed
-
- Burns E, Eliyahu T, Uematsu S, Akira S, Nussbaum G. TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J. Immunol. 2010;184:1455–1462. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
