

Repression of phosphoinositide-dependent protein kinase 1 expression by ciglitazone via Egr-1 represents a new approach for inhibition of lung cancer cell growth
- PMID: 24925061
- PMCID: PMC4061523
- DOI: 10.1186/1476-4598-13-149
Repression of phosphoinositide-dependent protein kinase 1 expression by ciglitazone via Egr-1 represents a new approach for inhibition of lung cancer cell growth
Abstract
Background: Peroxisome proliferator-activated receptors gamma (PPARγ) ligands have been shown to inhibit the growth of non-small cell lung cancer (NSCLC) cells. However, the mechanisms underlying this effect remain incompletely elucidated.
Methods: Cell proliferation and apoptosis were measured by cell viability, MTT and caspase3/7 activity assays. Phosphorylation/protein expression and gene silence/overexpression of AMPKα, phosphoinositide-dependent protein kinase 1 (PDK1), Egr-1 and PPARγ were performed by Western blot and siRNA/transfection assays. Dual-Luciferase Reporter Kit was used to measure the PPAR response elements (PPRE) reporter and PDK1 promoter activities, and ChIP assay was used to detect the Egr-1 protein binding to the DNA site in the PDK1 gene promoter.
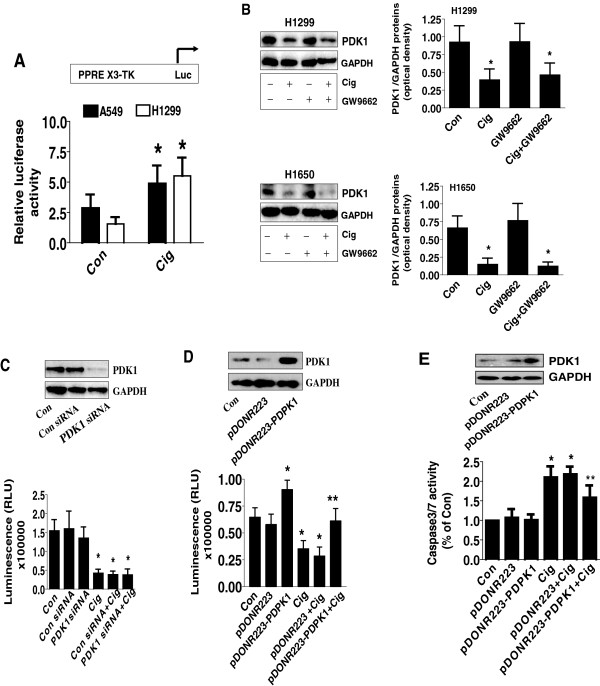
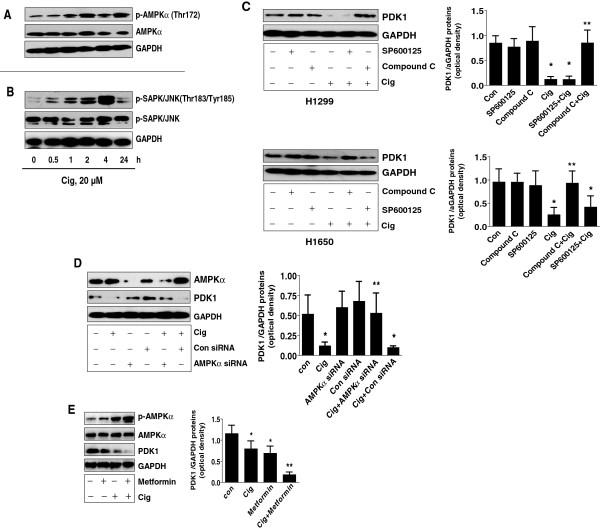
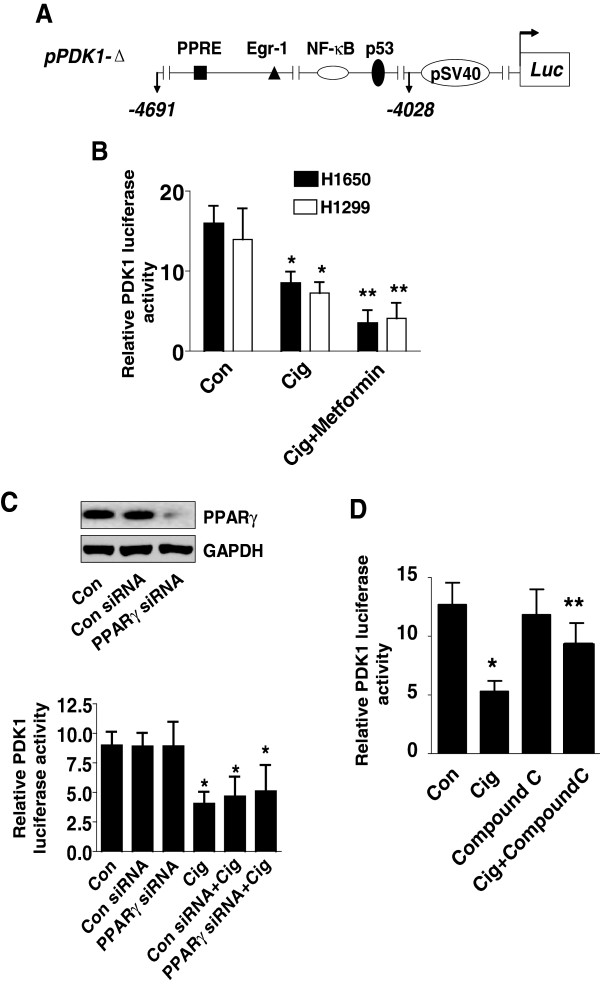
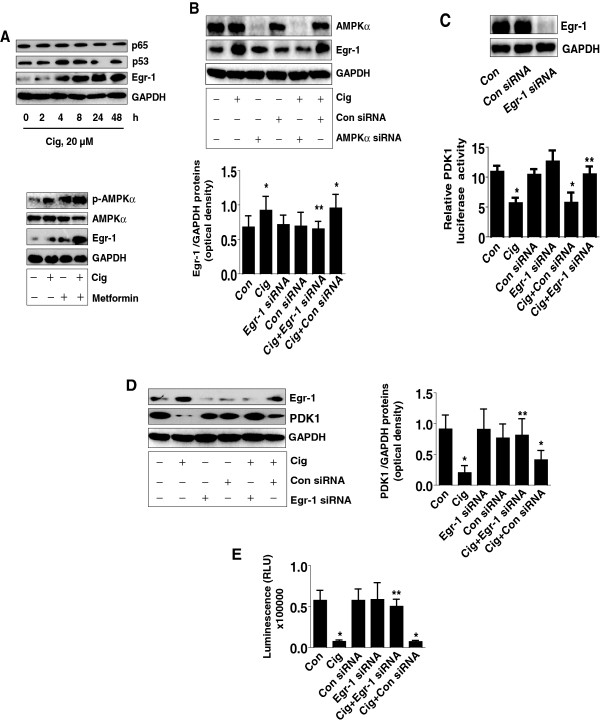
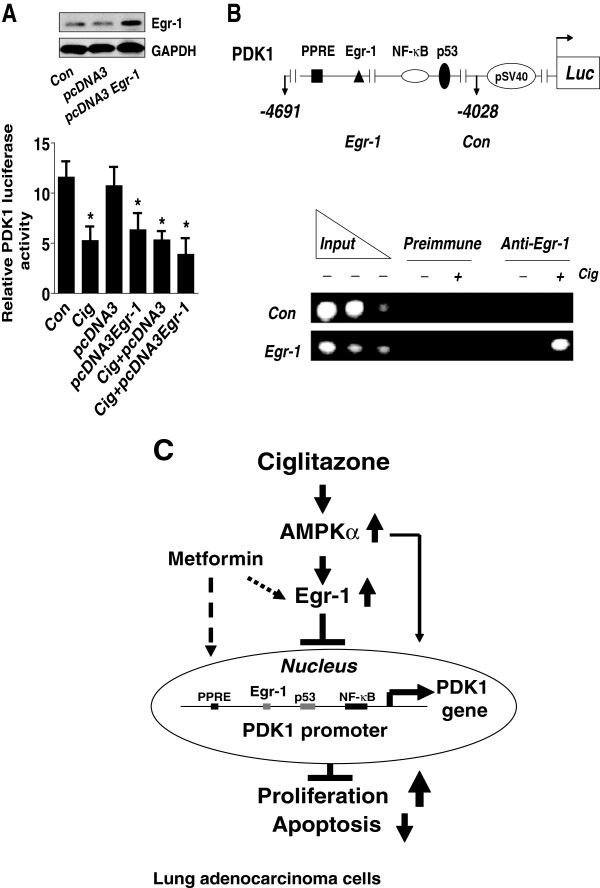
Results: We found that ciglitazone, one synthetic PPARγ ligand, inhibited growth and induced apoptosis of NSCLC cells through decreased expression of PDK1, which was not blocked by GW9662 (a specific PPARγ antagonist). Overexpression of PDK1 overcame the effect of ciglitazone on cell growth and caspase 3/7 activity. Ciglitazone increased the phosphorylation of AMPKα and c-Jun N-terminal kinase (JNK), and the inhibitor of AMPK (compound C), but not JNK (SP600125), reversed the effect of ciglitazone on PDK1 protein expression. Ciglitazone reduced PDK1 gene promoter activity, which was not observed in cells exposed to compound C, but not silenced of PPARγ siRNA. Combination of ciglitazone and metformin further reduced PDK1 expression and promoter activity. Furthermore, we showed that ciglitazone induced the protein expression of Egr-1, which was not observed in cells silencing of AMPKα. Moreover, silencing of Egr-1 abrogated the effect of ciglitazone on PDK1 promoter activity and cell growth. On the contrary, overexpression of Egr-1 enhanced the effect of ciglitazone on PDK1 gene promoter activity. ChIP assays demonstrated that ciglitazone induced Egr-1 protein bind to the specific DNA site in the PDK1 gene promoter.
Conclusion: Collectively, our results demonstrate that ciglitazone inhibits PDK1 expression through AMPKα-mediated induction of Egr-1 and Egr-1 binding to the specific DNA site in the PDK1 gene promoter, which is independent of PPARγ. Activation of AMPKα by metformin enhances the effect of ciglitazone. In turn, this leads to inhibition of NSCLC cell proliferation.
Figures

Similar articles
-
GW1929 inhibits α7 nAChR expression through PPARγ-independent activation of p38 MAPK and inactivation of PI3-K/mTOR: The role of Egr-1.Cell Signal. 2014 Apr;26(4):730-9. doi: 10.1016/j.cellsig.2013.12.019. Epub 2014 Jan 8. Cell Signal. 2014. PMID: 24412748
-
Targeting 3-phosphoinositide-dependent protein kinase 1 by N-acetyl-cysteine through activation of peroxisome proliferators activated receptor alpha in human lung cancer cells, the role of p53 and p65.J Exp Clin Cancer Res. 2013 Jul 18;32(1):43. doi: 10.1186/1756-9966-32-43. J Exp Clin Cancer Res. 2013. PMID: 23867003 Free PMC article.
-
Emodin Increases Expression of Insulin-Like Growth Factor Binding Protein 1 through Activation of MEK/ERK/AMPKα and Interaction of PPARγ and Sp1 in Lung Cancer.Cell Physiol Biochem. 2017;41(1):339-357. doi: 10.1159/000456281. Epub 2017 Jan 26. Cell Physiol Biochem. 2017. PMID: 28214826
-
Up-regulation of p21 gene expression by peroxisome proliferator-activated receptor gamma in human lung carcinoma cells.Clin Cancer Res. 2004 Mar 15;10(6):1911-9. doi: 10.1158/1078-0432.ccr-03-0985. Clin Cancer Res. 2004. PMID: 15041706
-
The Landscape of PDK1 in Breast Cancer.Cancers (Basel). 2022 Feb 5;14(3):811. doi: 10.3390/cancers14030811. Cancers (Basel). 2022. PMID: 35159078 Free PMC article. Review.
Cited by
-
PPARγ Modulators in Lung Cancer: Molecular Mechanisms, Clinical Prospects, and Challenges.Biomolecules. 2024 Feb 4;14(2):190. doi: 10.3390/biom14020190. Biomolecules. 2024. PMID: 38397426 Free PMC article. Review.
-
Cis-Regulation of an m6A Eraser by an Insertion Variant Associated with Survival of Patients With Non-Small Cell Lung Carcinoma.Adv Sci (Weinh). 2025 Feb;12(5):e2407652. doi: 10.1002/advs.202407652. Epub 2024 Dec 16. Adv Sci (Weinh). 2025. PMID: 39680684 Free PMC article.
-
Dual subcellular compartment delivery of doxorubicin to overcome drug resistant and enhance antitumor activity.Sci Rep. 2015 Nov 4;5:16125. doi: 10.1038/srep16125. Sci Rep. 2015. PMID: 26530454 Free PMC article.
-
Cell death and restoration of TRAIL-sensitivity by ciglitazone in resistant cervical cancer cells.Oncotarget. 2017 Nov 22;8(64):107744-107762. doi: 10.18632/oncotarget.22632. eCollection 2017 Dec 8. Oncotarget. 2017. PMID: 29296202 Free PMC article.
-
Ursolic acid inhibited growth of hepatocellular carcinoma HepG2 cells through AMPKα-mediated reduction of DNA methyltransferase 1.Mol Cell Biochem. 2015 Apr;402(1-2):63-74. doi: 10.1007/s11010-014-2314-x. Epub 2014 Dec 30. Mol Cell Biochem. 2015. PMID: 25547067
References
-
- Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, Kuruvilla DS, Shin Y, He Y, Bruning JB, Marciano DP, Cameron MD, Laznik D, Jurczak MJ, Schürer SC, Vidović D, Shulman GI, Spiegelman BM, Griffin PR. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
