

TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy
- PMID: 24925317
- PMCID: PMC4189907
- DOI: 10.1093/hmg/ddu297
TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy
Abstract
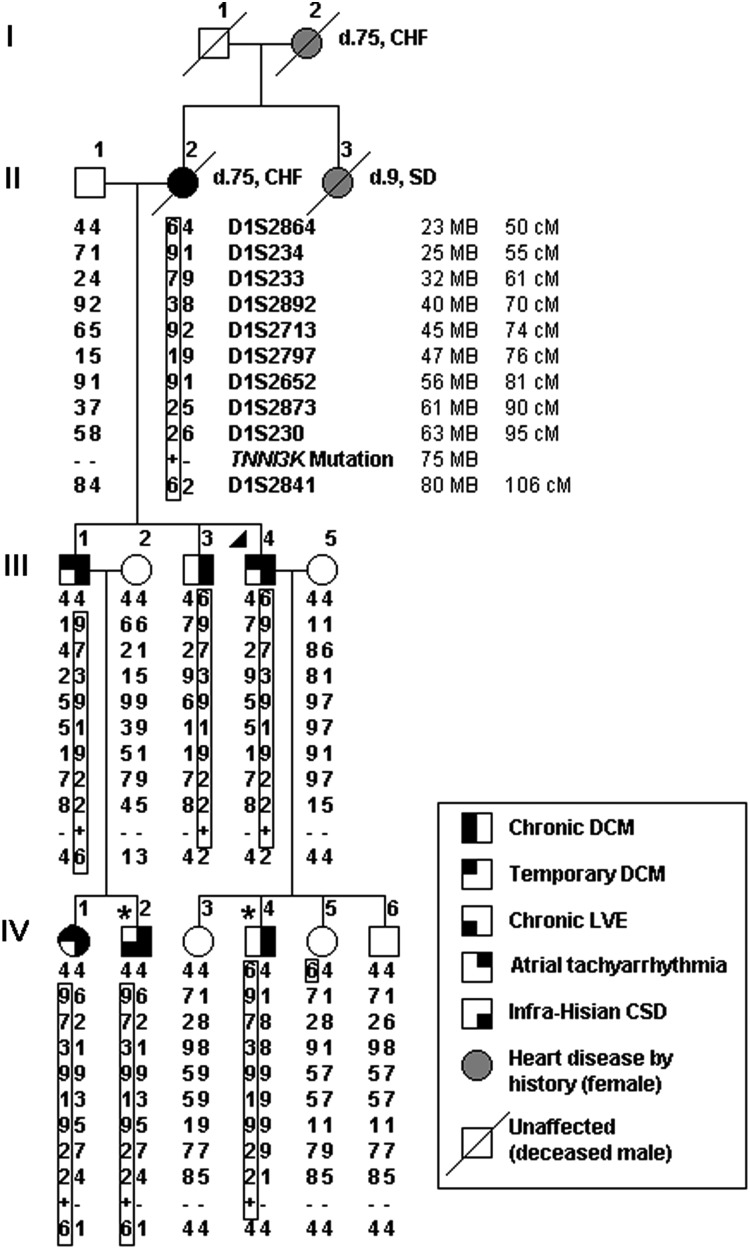
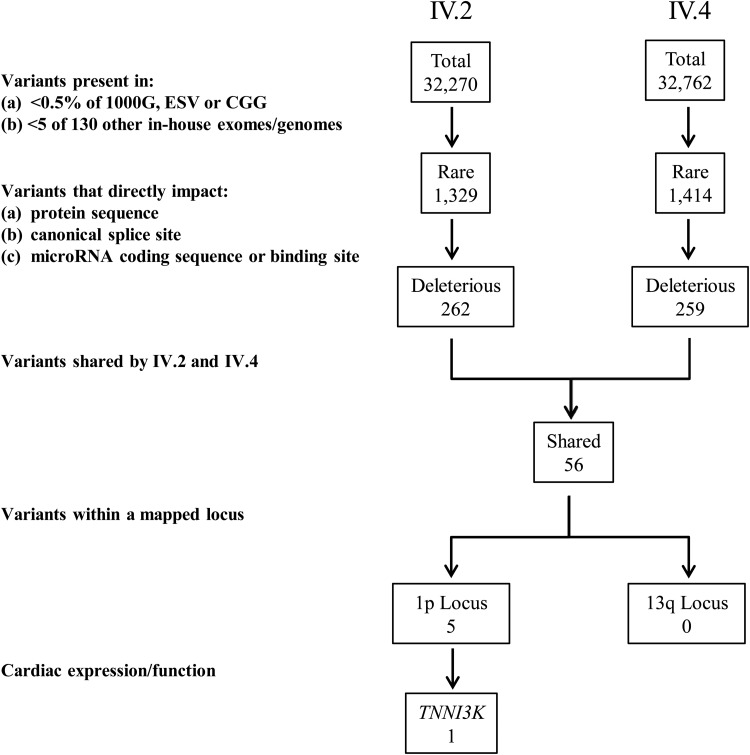
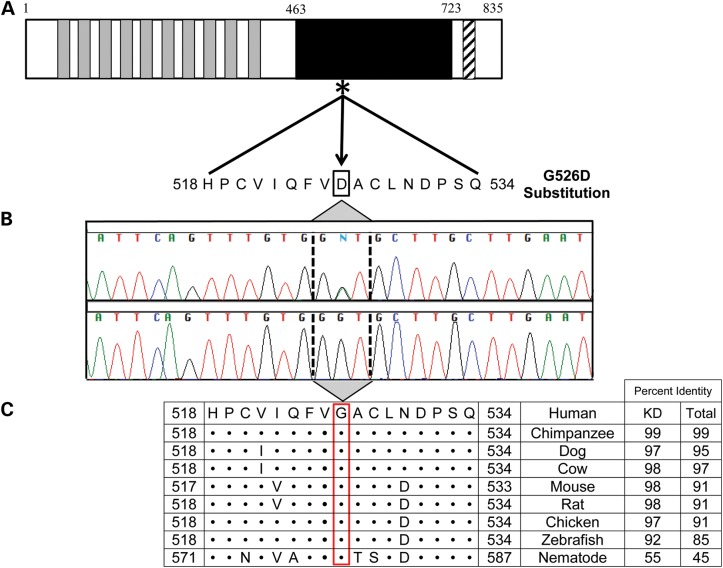
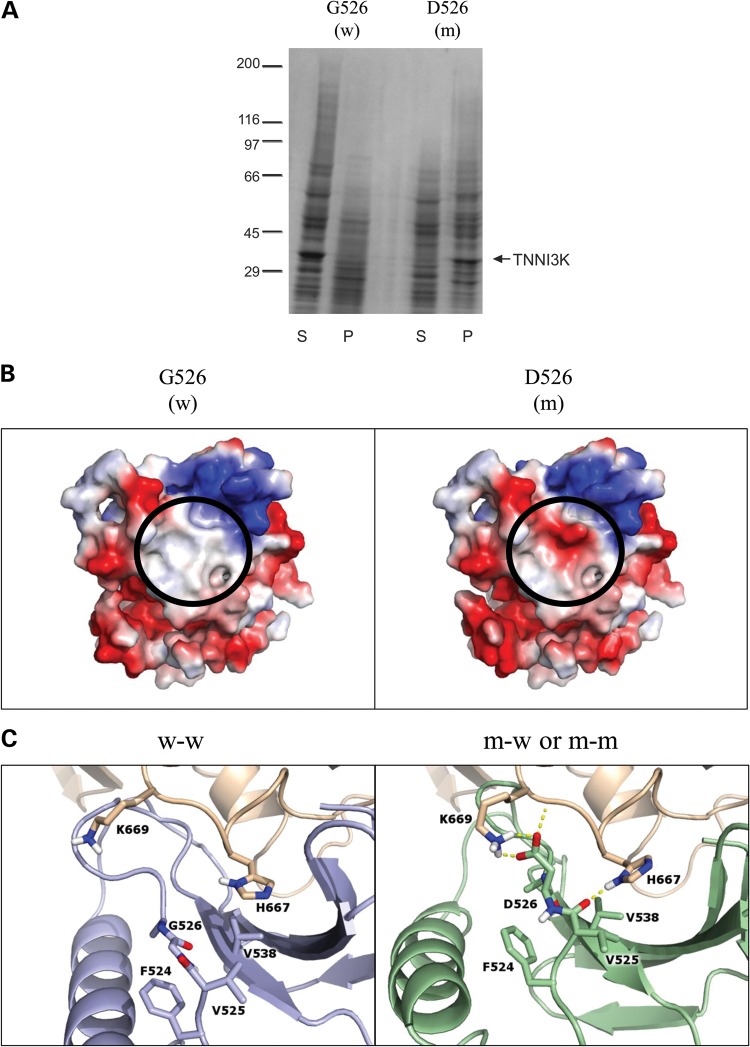
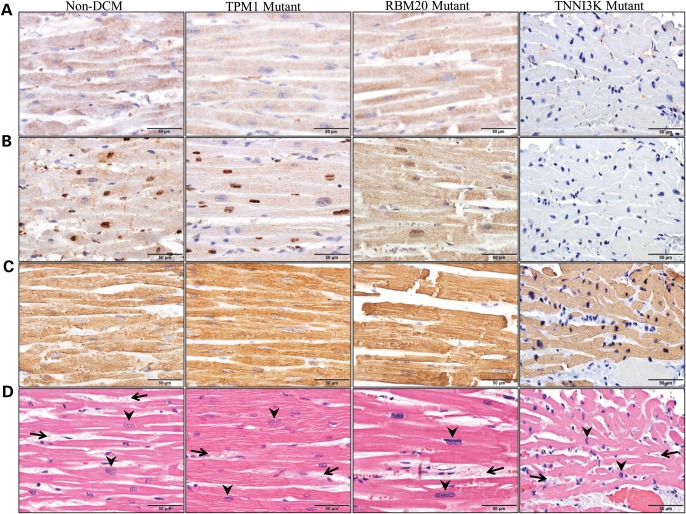
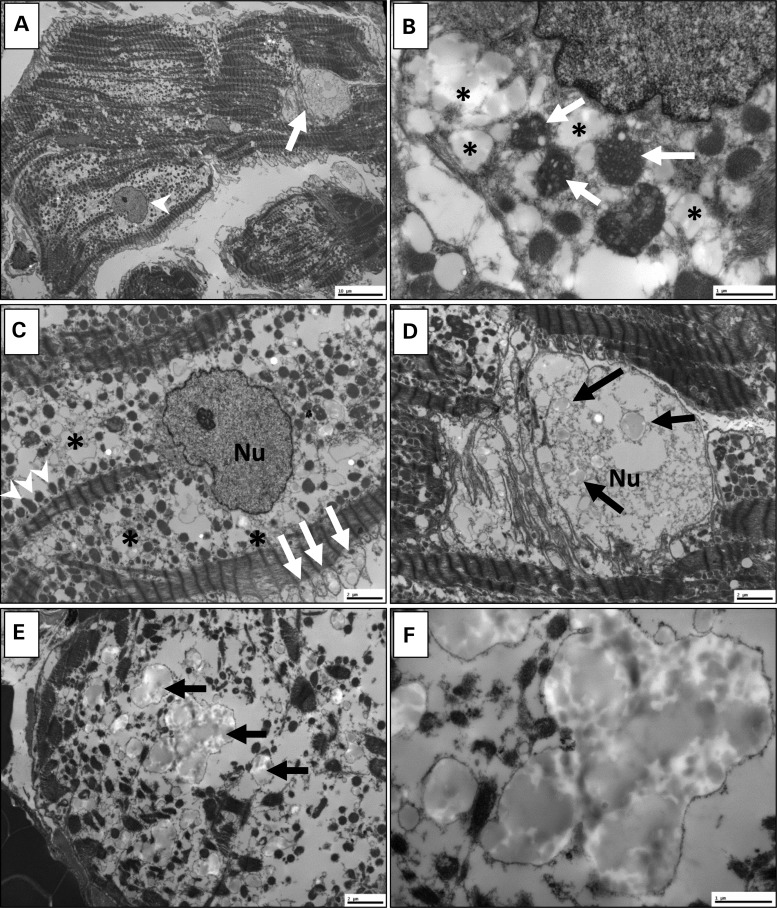
Locus mapping has uncovered diverse etiologies for familial atrial fibrillation (AF), dilated cardiomyopathy (DCM), and mixed cardiac phenotype syndromes, yet the molecular basis for these disorders remains idiopathic in most cases. Whole-exome sequencing (WES) provides a powerful new tool for familial disease gene discovery. Here, synergistic application of these genomic strategies identified the pathogenic mutation in a familial syndrome of atrial tachyarrhythmia, conduction system disease (CSD), and DCM vulnerability. Seven members of a three-generation family exhibited the variably expressed phenotype, three of whom manifested CSD and clinically significant arrhythmia in childhood. Genome-wide linkage analysis mapped two equally plausible loci to chromosomes 1p3 and 13q12. Variants from WES of two affected cousins were filtered for rare, predicted-deleterious, positional variants, revealing an unreported heterozygous missense mutation disrupting the highly conserved kinase domain in TNNI3K. The G526D substitution in troponin I interacting kinase, with the most deleterious SIFT and Polyphen2 scores possible, resulted in abnormal peptide aggregation in vitro and in silico docking models predicted altered yet energetically favorable wild-type mutant dimerization. Ventricular tissue from a mutation carrier displayed histopathological hallmarks of DCM and reduced TNNI3K protein staining with unique amorphous nuclear and sarcoplasmic inclusions. In conclusion, mutation of TNNI3K, encoding a heart-specific kinase previously shown to modulate cardiac conduction and myocardial function in mice, underlies a familial syndrome of electrical and myopathic heart disease. The identified substitution causes a TNNI3K aggregation defect and protein deficiency, implicating a dominant-negative loss of function disease mechanism.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Lloyd-Jones D.M., Wang T.J., Leip E.P., Larson M.G., Levy D., Vasan R.S., D'Agostino R.B., Massaro J.M., Beiser A., Wolf P.A., et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110:1042–1046. - PubMed
-
- Darbar D., Herron K.J., Ballew J.D., Jahangir A., Gersh B.J., Shen W.-K., Hammill S.C., Packer D.L., Olson T.M. Familial atrial fibrillation is a genetically heterogeneous disorder. J. Am. Coll. Cardiol. 2003;41:2185–2192. - PubMed
-
- Chen Y.H., Xu S.J., Bendahhou S., Wang X.L., Wang Y., Xu W.Y., Jin H.W., Sun H., Su X.Y., Zhuang Q.N., et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. - PubMed
-
- Zhang X., Chen S., Yoo S., Chakrabarti S., Zhang T., Ke T., Oberti C., Yong S.L., Fang F., Li L., et al. Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell. 2008;135:1017–1027. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
