

Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads
- PMID: 24925680
- PMCID: PMC4074385
- DOI: 10.1186/1471-2105-15-182
Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads
Abstract
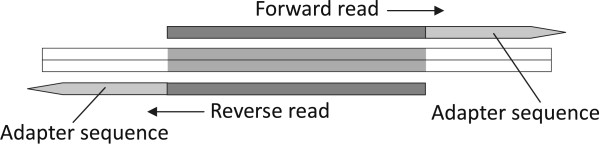
Background: Adapter trimming is a prerequisite step for analyzing next-generation sequencing (NGS) data when the reads are longer than the target DNA/RNA fragments. Although typically used in small RNA sequencing, adapter trimming is also used widely in other applications, such as genome DNA sequencing and transcriptome RNA/cDNA sequencing, where fragments shorter than a read are sometimes obtained because of the limitations of NGS protocols. For the newly emerged Nextera long mate-pair (LMP) protocol, junction adapters are located in the middle of all properly constructed fragments; hence, adapter trimming is essential to gain the correct paired reads. However, our investigations have shown that few adapter trimming tools meet both efficiency and accuracy requirements simultaneously. The performances of these tools can be even worse for paired-end and/or mate-pair sequencing.
Results: To improve the efficiency of adapter trimming, we devised a novel algorithm, the bit-masked k-difference matching algorithm, which has O(kn) expected time with O(m) space, where k is the maximum number of differences allowed, n is the read length, and m is the adapter length. This algorithm makes it possible to fully enumerate all candidates that meet a specified threshold, e.g. error ratio, within a short period of time. To improve the accuracy of this algorithm, we designed a simple and easy-to-explain statistical scoring scheme to evaluate candidates in the pattern matching step. We also devised scoring schemes to fully exploit the paired-end/mate-pair information when it is applicable. All these features have been implemented in an industry-standard tool named Skewer (https://sourceforge.net/projects/skewer). Experiments on simulated data, real data of small RNA sequencing, paired-end RNA sequencing, and Nextera LMP sequencing showed that Skewer outperforms all other similar tools that have the same utility. Further, Skewer is considerably faster than other tools that have comparative accuracies; namely, one times faster for single-end sequencing, more than 12 times faster for paired-end sequencing, and 49% faster for LMP sequencing.
Conclusions: Skewer achieved as yet unmatched accuracies for adapter trimming with low time bound.
Figures
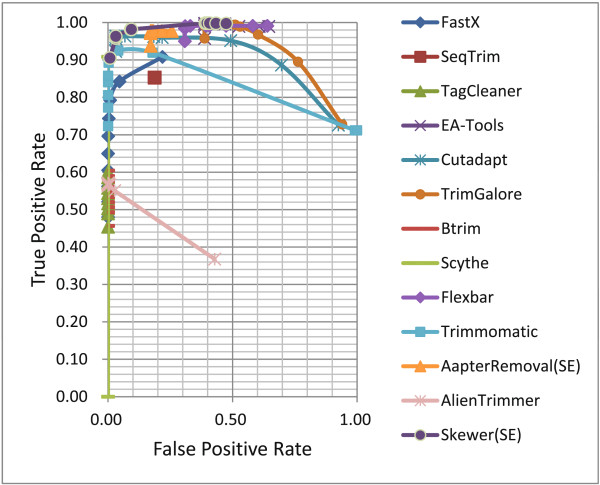
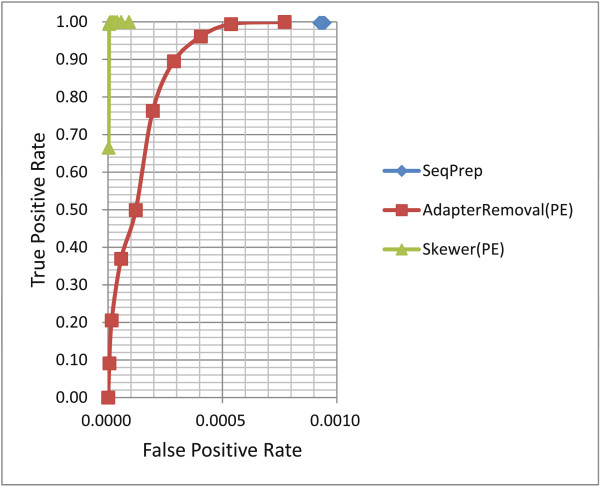
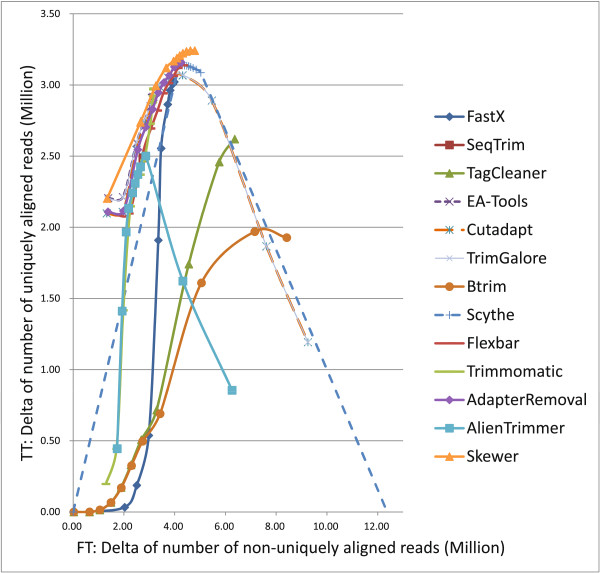
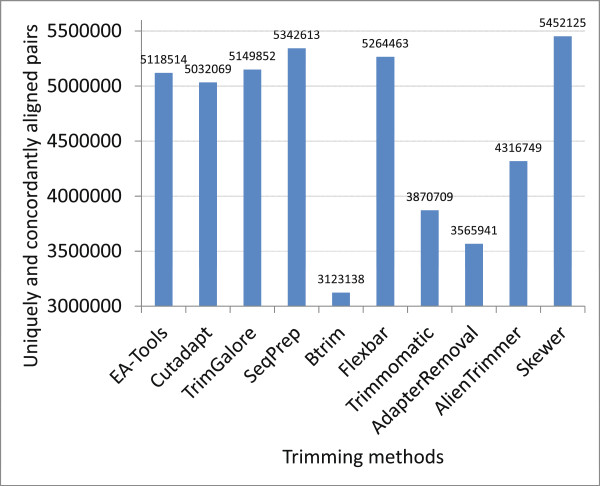
References
-
- Smith TF, Waterman MS. Identification of common molecular subsequences . J Mol Biol. 1981;147(1):195–197. - PubMed
-
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads . EMBnet.journal. 2011;17:10–12.
-
- Ukkonen E. Finding approximate patterns in strings . J Algorithm. 1985;6(1):132–137.
-
- Myers G. A fast bit-vector algorithm for approximate string matching based on dynamic programming . J ACM. 1999;46(3):395–415.
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
