

Altered histone modifications in gliomas
- PMID: 24926467
- PMCID: PMC4049557
- DOI: 10.14791/btrt.2014.2.1.7
Altered histone modifications in gliomas
Abstract
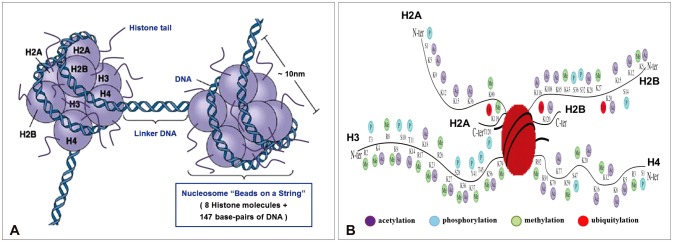
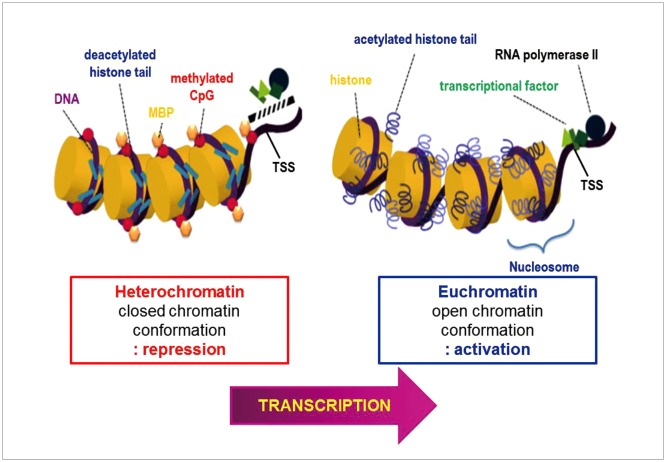
Gliomas are the most frequently occurring primary brain tumors in adults. Although they exist in different malignant stages, including histologically benign forms and highly aggressive states, most gliomas are clinically challenging for neuro-oncologists because of their infiltrative growth patterns and inherent relapse tendency with increased malignancy. Once this disease reaches the glioblastoma multiforme stage, the prognosis of patients is dismal: median survival time is 15 months. Extensive genetic analyses of glial tumors have revealed a variety of deregulated genetic pathways involved in DNA repair, apoptosis, cell migration/adhesion, and cell cycle. Recently, it has become evident that epigenetic alterations may also be an important factor for glioma genesis. Of epigenetic marks, histone modification is a key mark that regulates gene expression and thus modulates a wide range of cellular processes. In this review, I discuss the neuro-oncological significance of altered histone modifications and modifiers in glioma patients while briefly overviewing the biological roles of histone modifications.
Keywords: Acetylation; Epigenetics; Glioblastoma; Glioma; Histone; Methylation.
Conflict of interest statement
The author has no financial conflicts of interest.
Figures




References
-
- Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. Nat Genet. 1995;10:20–27. - PubMed
-
- Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219–220. - PubMed
-
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. - PubMed
-
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
