

Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis
- PMID: 24927234
- PMCID: PMC4226057
- DOI: 10.1164/rccm.201404-0703OC
Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis
Abstract
Rationale: Ivacaftor is a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator recently approved for patients with CF age 6 and older with the G551D mutation.
Objectives: To evaluate ivacaftor in a postapproval setting and determine mechanism of action and response of clinically relevant markers.
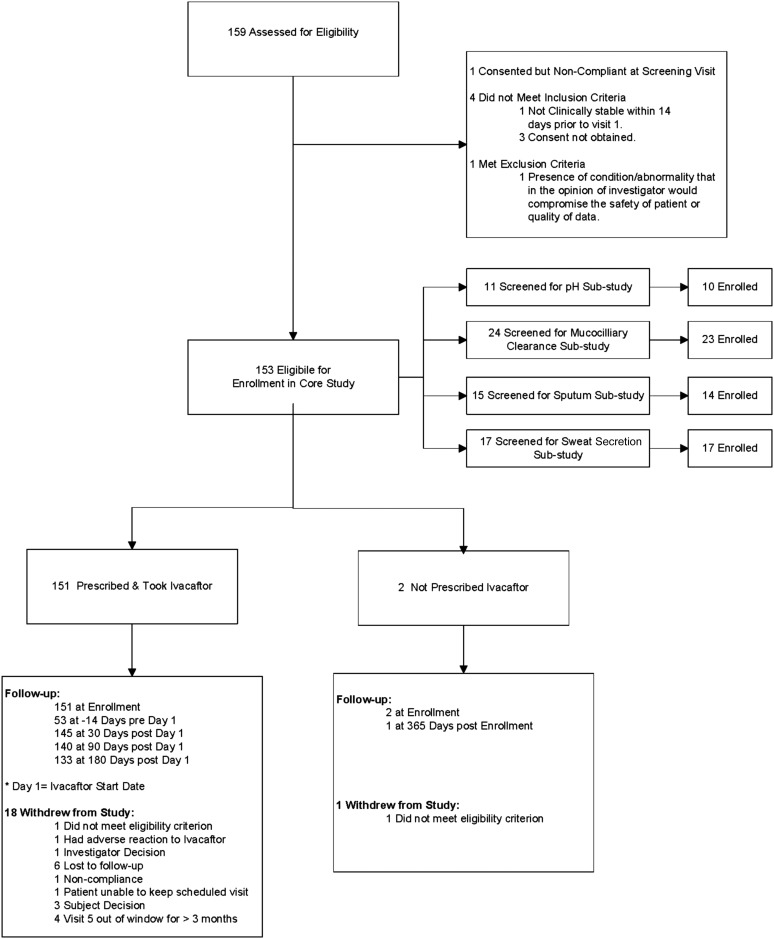
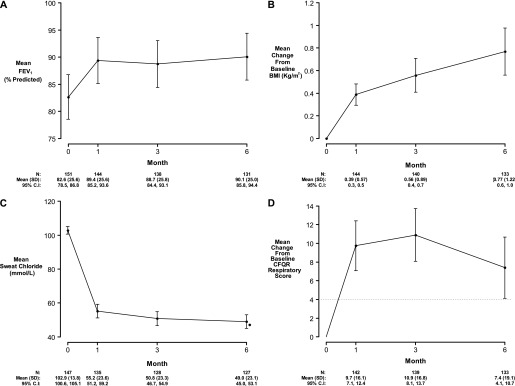
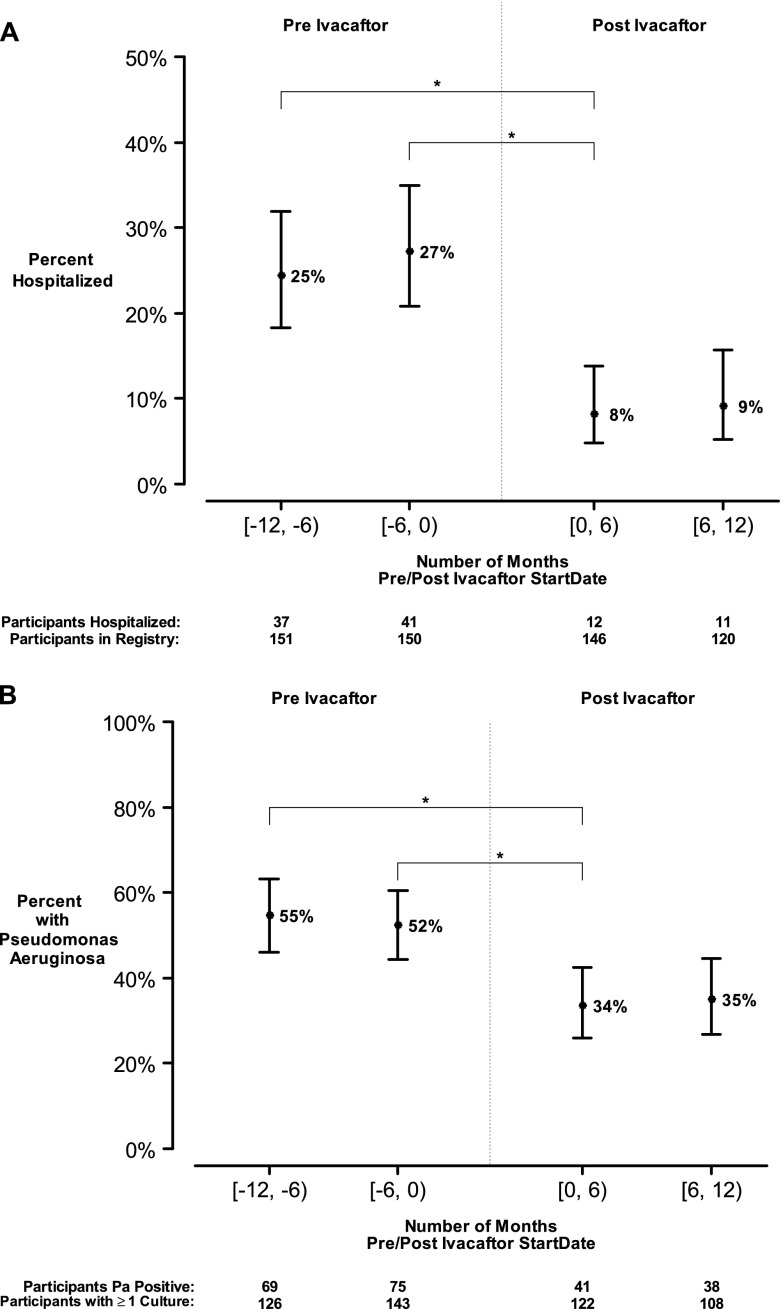
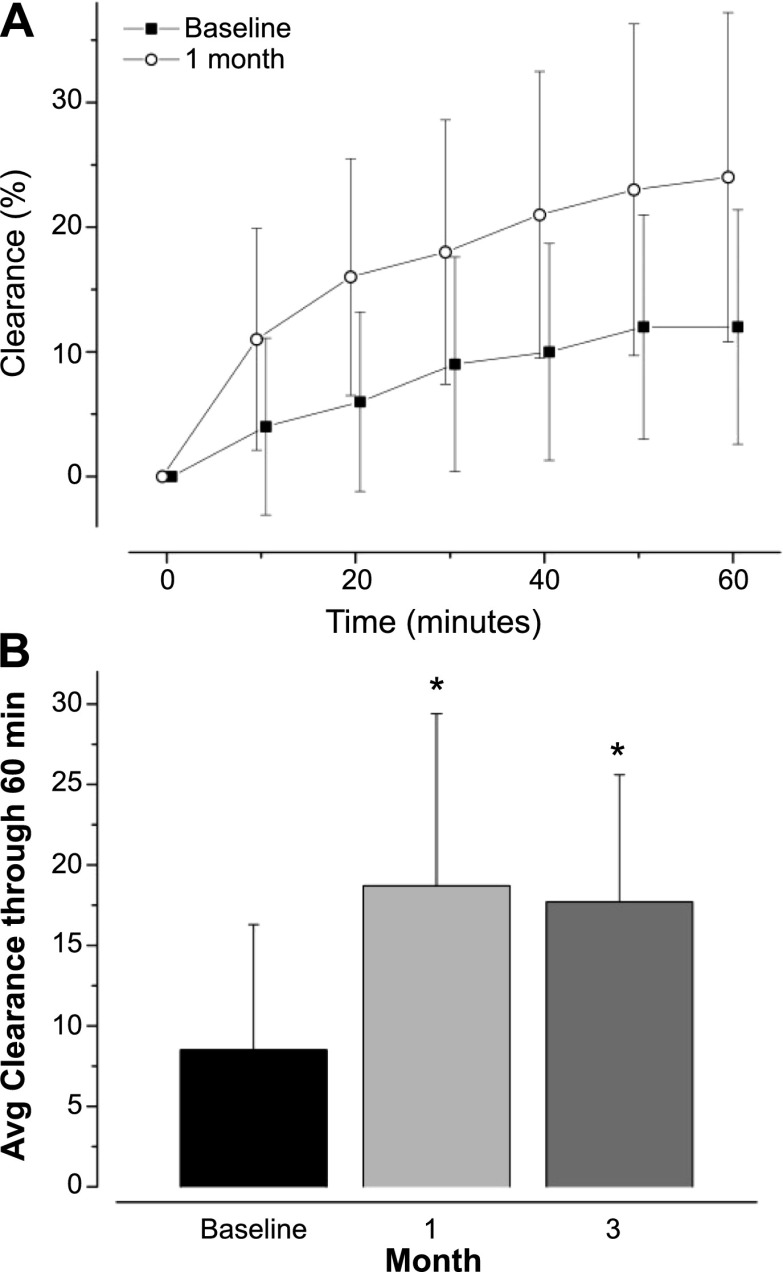
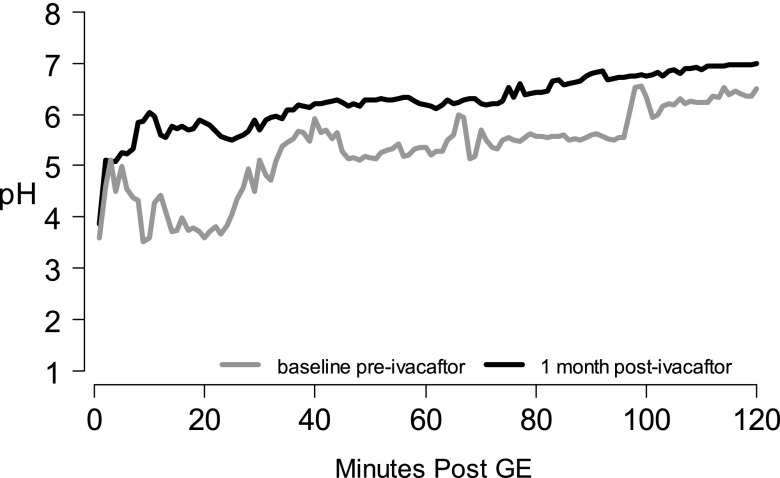
Methods: We conducted a longitudinal cohort study in 2012-2013 in G551D CF patients age 6 and older with no prior exposure to ivacaftor. Study assessments were performed at baseline, 1, 3, and 6 months after ivacaftor initiation. Substudies evaluated mucociliary clearance, β-adrenergic sweat secretion rate, gastrointestinal pH, and sputum inflammation and microbiology Measurements and Main Results: A total of 151 of 153 subjects were prescribed ivacaftor and 88% completed the study through 6 months. FEV1 % predicted improved from baseline to 6 months (mean absolute change, 6.7%; P < 0.001). Similarly, body mass index improved from baseline to 6 months (mean change, 0.8 kg/m(2); P < 0.001). Sweat chloride decreased from baseline to 6 months (mean change, -53.8 mmol/L; 95% confidence interval, -57.7 to -49.9; P < 0.001), reflecting augmented CFTR function. There was significant improvement in hospitalization rate (P < 0.001) and Pseudomonas aeruginosa burden (P < 0.01). Significant improvements in mucociliary clearance (P < 0.001), gastrointestinal pH (P = 0.001), and microbiome were also observed, providing clinical mechanisms underlying the therapeutic benefit of ivacaftor.
Conclusions: Significant clinical and physiologic improvements were observed on initiation of ivacaftor in a broad patient population, including reduced infection with P. aeruginosa. Biomarker studies substantially improve the understanding of the mechanistic consequences of CFTR modulation on pulmonary and gastrointestinal physiology.
Keywords: CFTR modulator; Pseudomonas aeruginosa; cystic fibrosis; ivacaftor; pH.
Figures
Comment in
-
Ivacaftor: from bench to bedside... and back again.Am J Respir Crit Care Med. 2014 Jul 15;190(2):128-9. doi: 10.1164/rccm.201406-1122ED. Am J Respir Crit Care Med. 2014. PMID: 25025350 No abstract available.
References
-
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. - PubMed
-
- Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689. - PubMed
-
- Gelfond D, Borowitz D.Gastrointestinal complications of cystic fibrosis Clin Gastroenterol Hepatol 201311333–342.quiz e330–331 - PubMed
-
- Collie JT, Massie RJ, Jones OA, Legrys VA, Greaves RF. Sixty-five years since the New York heat wave: advances in sweat testing for cystic fibrosis. Pediatr Pulmonol. 2014;49:106–117. - PubMed
-
- Cystic Fibrosis Foundation. Cystic Fibrosis Foundation National Patient Registry annual data report 2012. Bethesda, MD: Cystic Fibrosis Foundation; 2013
Publication types
MeSH terms
Substances
Grants and funding
- UL1TR000423/TR/NCATS NIH HHS/United States
- UL1TR001111/TR/NCATS NIH HHS/United States
- UL1 TR000165/TR/NCATS NIH HHS/United States
- P30 DK089507/DK/NIDDK NIH HHS/United States
- UL1 TR001082/TR/NCATS NIH HHS/United States
- UL1 TR001111/TR/NCATS NIH HHS/United States
- UL1TR000165/TR/NCATS NIH HHS/United States
- P30 DK072482/DK/NIDDK NIH HHS/United States
- UL1TR001082/TR/NCATS NIH HHS/United States
- DK072482/DK/NIDDK NIH HHS/United States
- DK089507/DK/NIDDK NIH HHS/United States
- KL2 TR000421/TR/NCATS NIH HHS/United States
- UL1 TR000423/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
