

Hsp90 modulates PPARγ activity in a mouse model of nonalcoholic fatty liver disease
- PMID: 24927728
- PMCID: PMC4109764
- DOI: 10.1194/jlr.M048918
Hsp90 modulates PPARγ activity in a mouse model of nonalcoholic fatty liver disease
Abstract
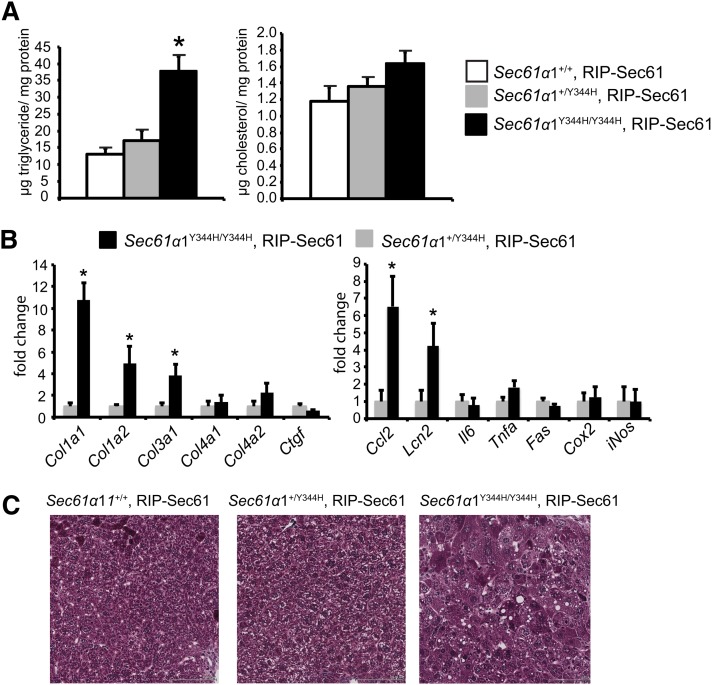
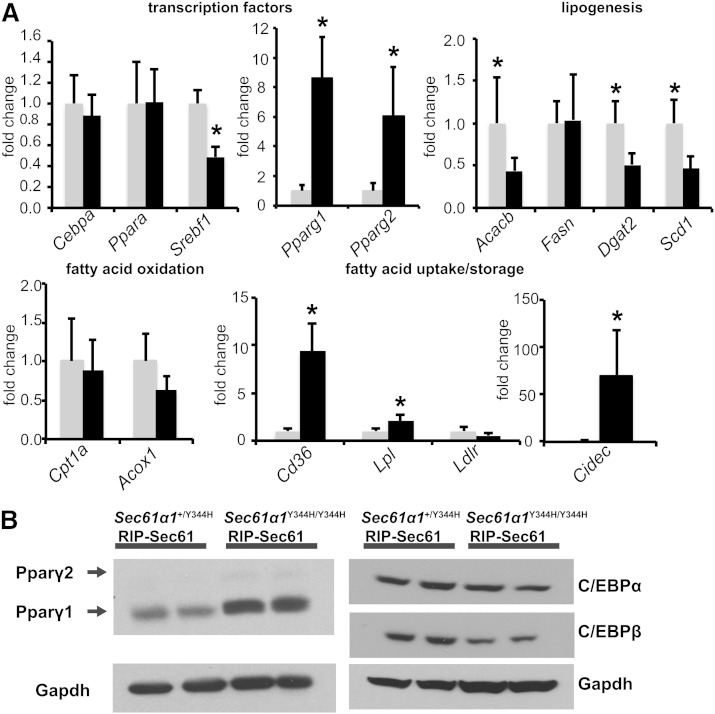
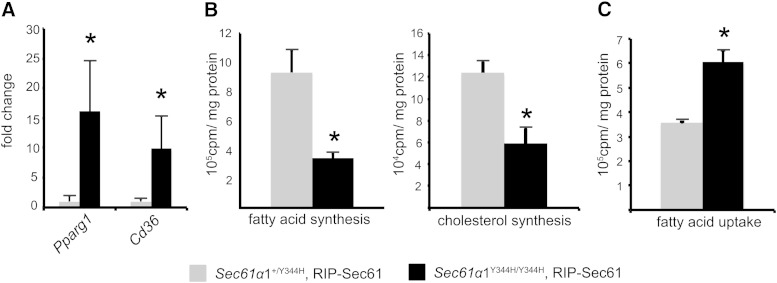
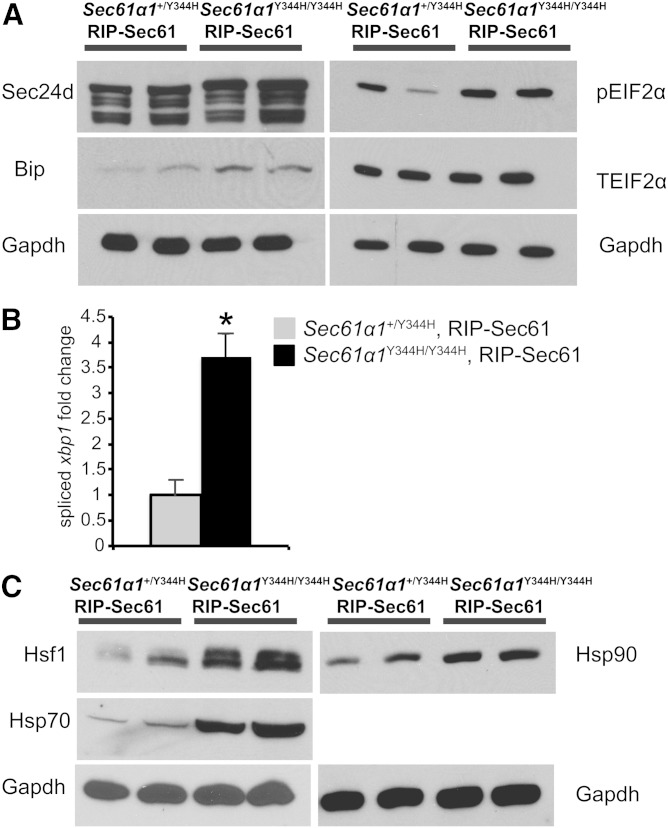
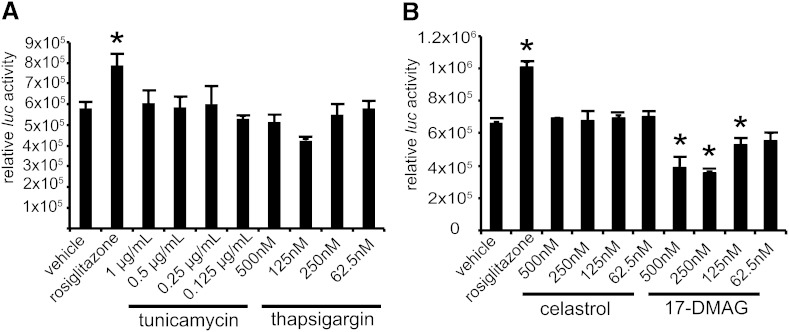
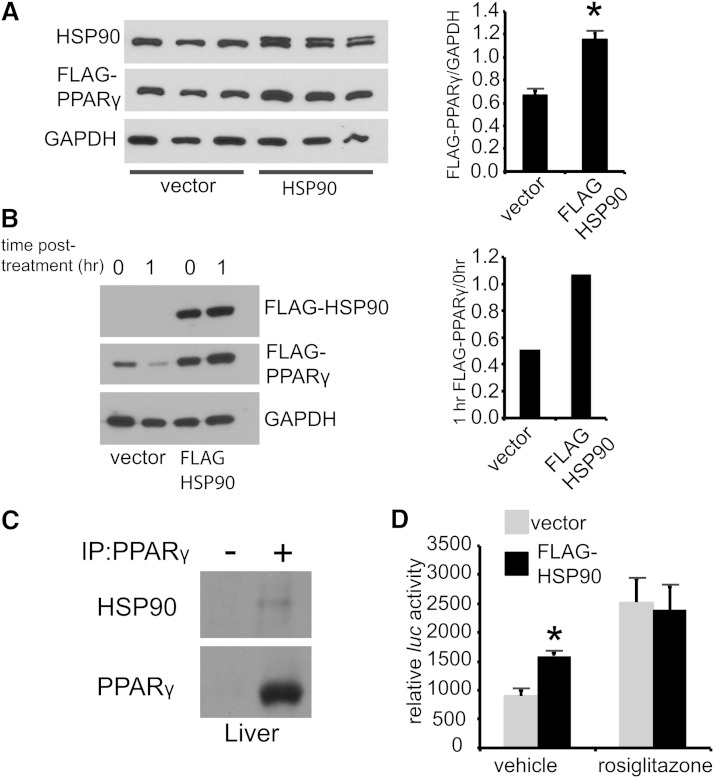
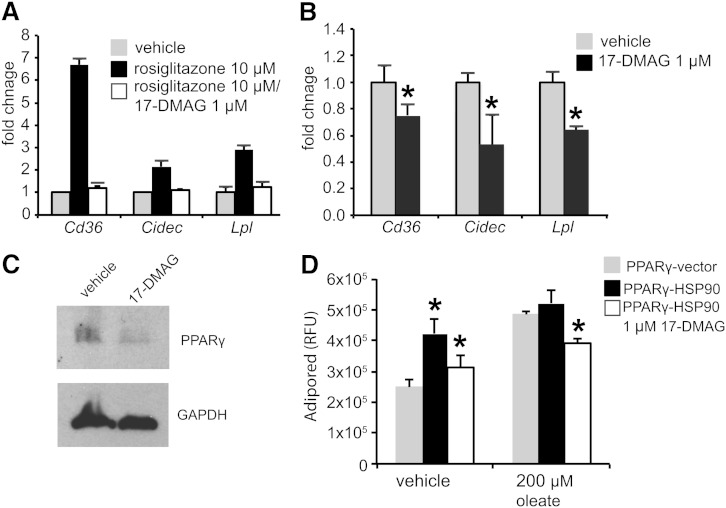
Nonalcoholic fatty liver disease (NAFLD) is a highly prevalent complication of obesity, yet cellular mechanisms that lead to its development are not well defined. Previously, we have documented hepatic steatosis in mice carrying a mutation in the Sec61a1 gene. Here we examined the mechanism behind NAFLD in Sec61a1 mutant mice. Livers of mutant mice exhibited upregulation of Pparg and its target genes Cd36, Cidec, and Lpl, correlating with increased uptake of fatty acid. Interestingly, these mice also displayed activation of the heat shock response (HSR), with elevated levels of heat shock protein (Hsp) 70, Hsp90, and heat shock factor 1. In cell lines, inhibition of Hsp90 function reduced Pparγ signaling and protein levels. Conversely, overexpression of Hsp90 increased Pparγ signaling and protein levels by reducing degradation. This may occur via a physical interaction as Hsp90 and Pparγ coimmunoprecipitated in vivo. Furthermore, inhibition of Hsp90 in Sec61a1 mutant hepatocytes also reduced Pparγ protein levels and signaling. Finally, overexpression of Hsp90 in liver cell lines increased neutral lipid accumulation, and this accumulation was blocked by Hsp90 inhibition. Our results show that the HSR and Hsp90 play an important role in the development of NAFLD, opening new avenues for the prevention and treatment of this highly prevalent disease.
Keywords: heat shock protein 90; heat shock response; peroxisome proliferator-activated receptor γ; steatosis.
Copyright © 2014 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Browning J. D., Szczepaniak L. S., Dobbins R., Nuremberg P., Horton J. D., Cohen J. C., Grundy S. M., Hobbs H. H. 2004. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 40: 1387–1395. - PubMed
-
- Targher G., Day C. P., Bonora E. 2010. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 363: 1341–1350. - PubMed
-
- Koonen D. P., Jacobs R. L., Febbraio M., Young M. E., Soltys C. L., Ong H., Vance D. E., Dyck J. R. 2007. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 56: 2863–2871. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
