

Pulmonary alveolar proteinosis in children on La Réunion Island: a new inherited disorder?
- PMID: 24927752
- PMCID: PMC4062771
- DOI: 10.1186/1750-1172-9-85
Pulmonary alveolar proteinosis in children on La Réunion Island: a new inherited disorder?
Abstract
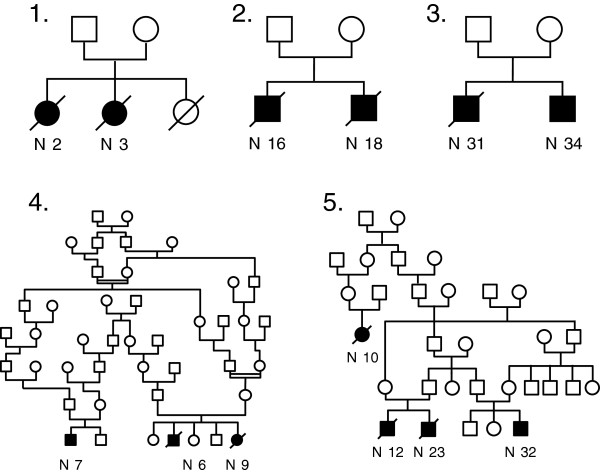
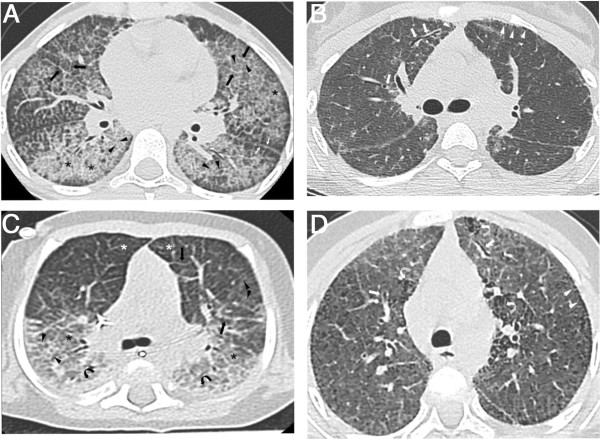
Background: Pulmonary alveolar proteinosis (PAP) is very rare in children. Only a few small series have been published, with little information about long-term progression. The objective of our study was to describe the clinical, radiological and pathological features, and the long-term course of PAP in a cohort of 34 children from La Réunion Island.
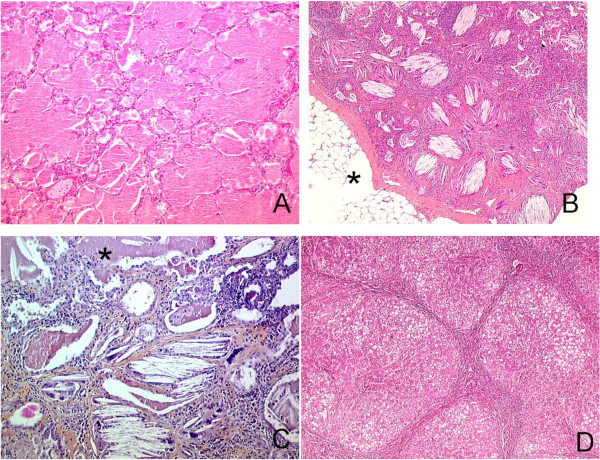
Methods: Data were retrospectively collected from medical files. Radiological and pathological elements were reviewed by two pediatric radiologists and three pathologists, respectively.
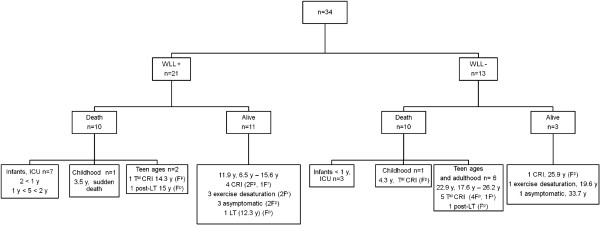
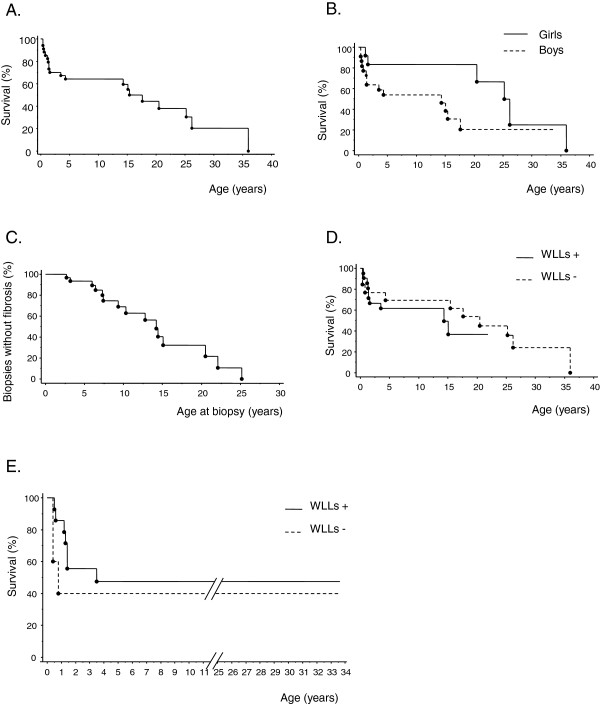
Results: Thirteen cases were familial and 32/34 (94%) cases were family connected. Disease onset occurred in the first six months of life in 82% of the patients. Thoracic computed tomography scans showed the typical "crazy-paving" pattern in 94% of cases. Respiratory disease was associated with a liver disorder, with the detection of liver enlargement at diagnosis in 56% of cases. The course of the disease was characterized by frequent progression to chronic respiratory insufficiency, accompanied by the appearance of cholesterol granulomas and pulmonary fibrosis. Overall prognosis was poor, with a mortality of 59% and an overall five-year survival rate from birth of 64%. Whole-lung lavages were performed in 21 patients, with no significant effect on survival. Liver disease progressed to cirrhosis in 18% of children, with no severe complication.
Conclusions: PAP in children from la Réunion Island is characterized by an early onset, associated liver involvement, poor prognosis and frequent progression to lung fibrosis, despite whole-lung lavages treatment. The geographic clustering of patients and the detection of many familial links between most of the cases strongly suggest a genetic etiology, with an autosomal recessive mode of inheritance.
Figures
References
-
- Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, Kasahara Y, Tatsumi K, Hojo M, Ichiwata T, Tanaka N, Yamaguchi E, Eda R, Oishi K, Tsuchihashi Y, Kaneko C, Nukiwa T, Sakatani M, Krischer JP, Nakata K. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177:752–762. doi: 10.1164/rccm.200708-1271OC. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
