

Uric acid and thiocyanate as competing substrates of lactoperoxidase
- PMID: 24928513
- PMCID: PMC4139211
- DOI: 10.1074/jbc.M113.544957
Uric acid and thiocyanate as competing substrates of lactoperoxidase
Abstract
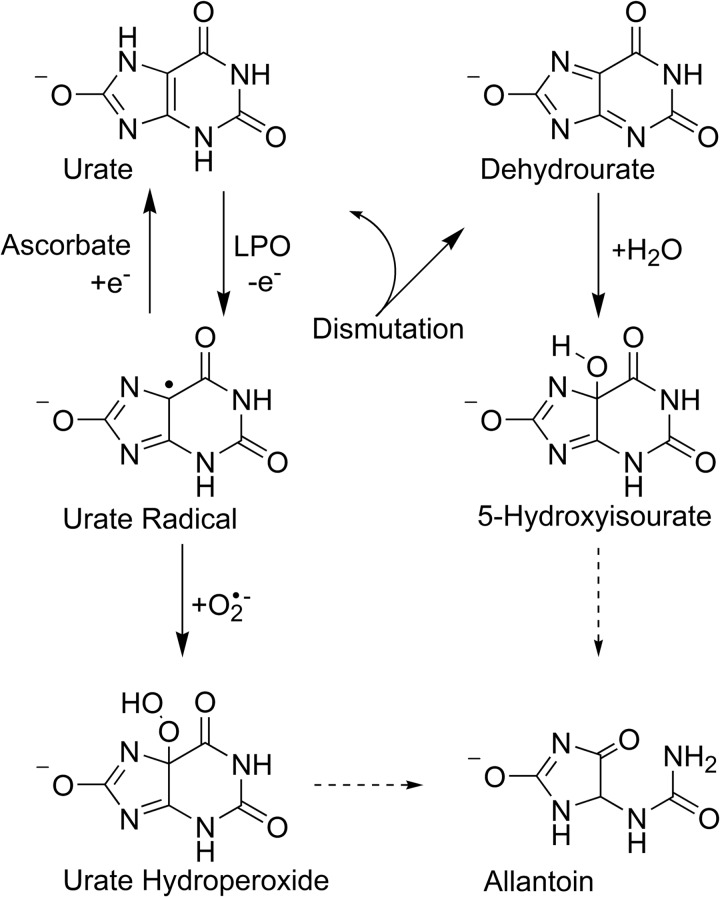
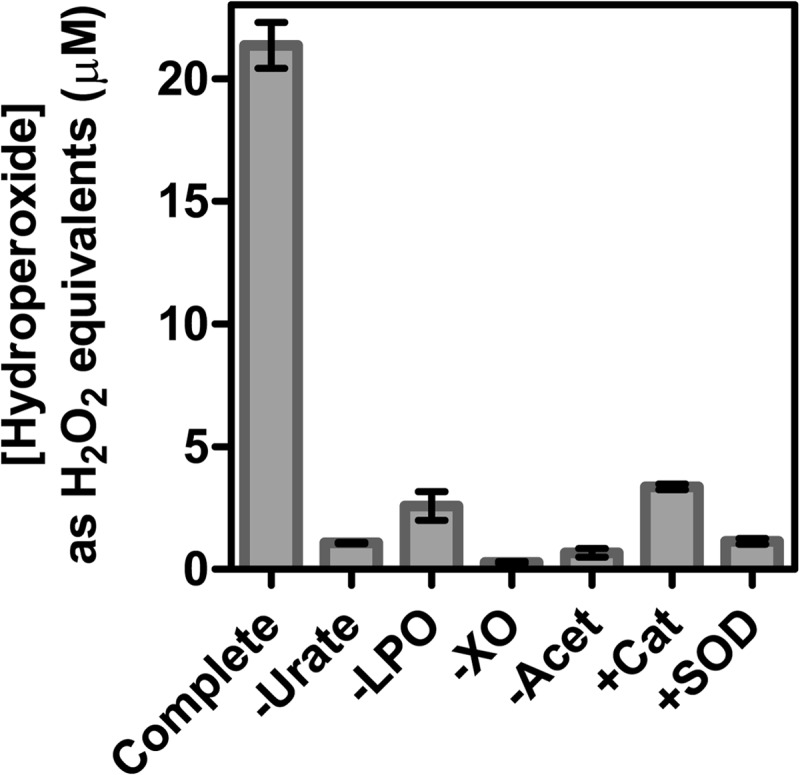
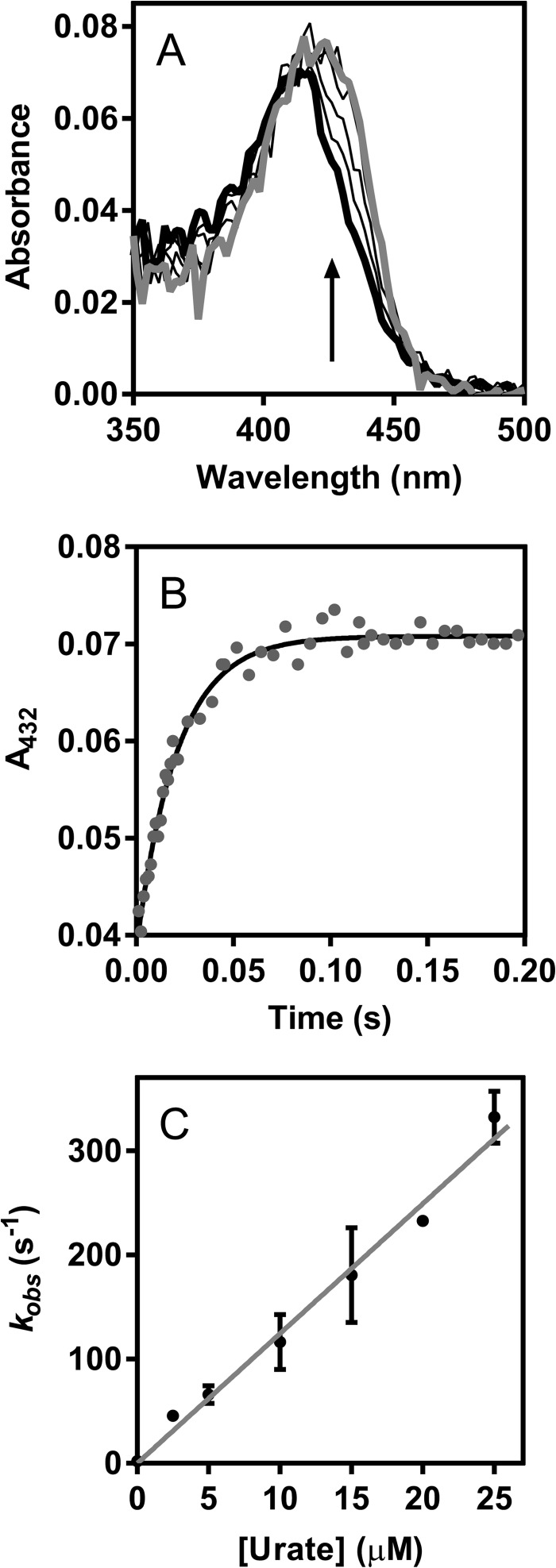
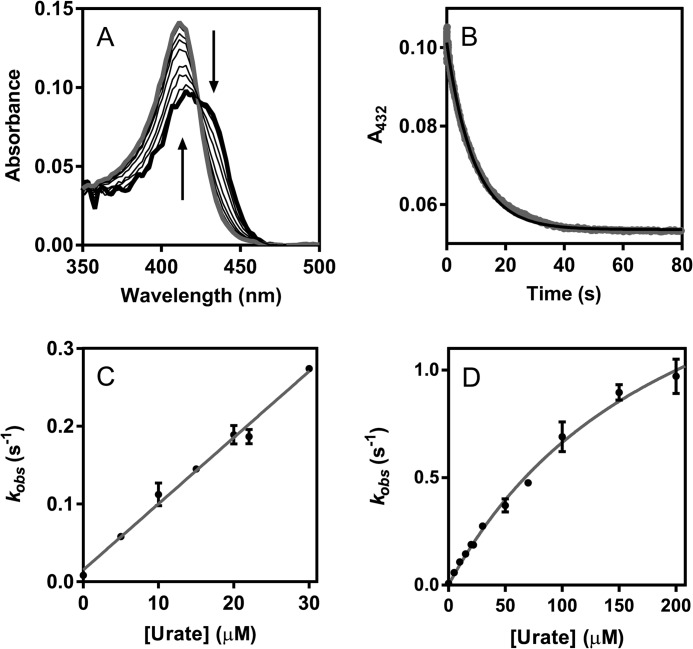
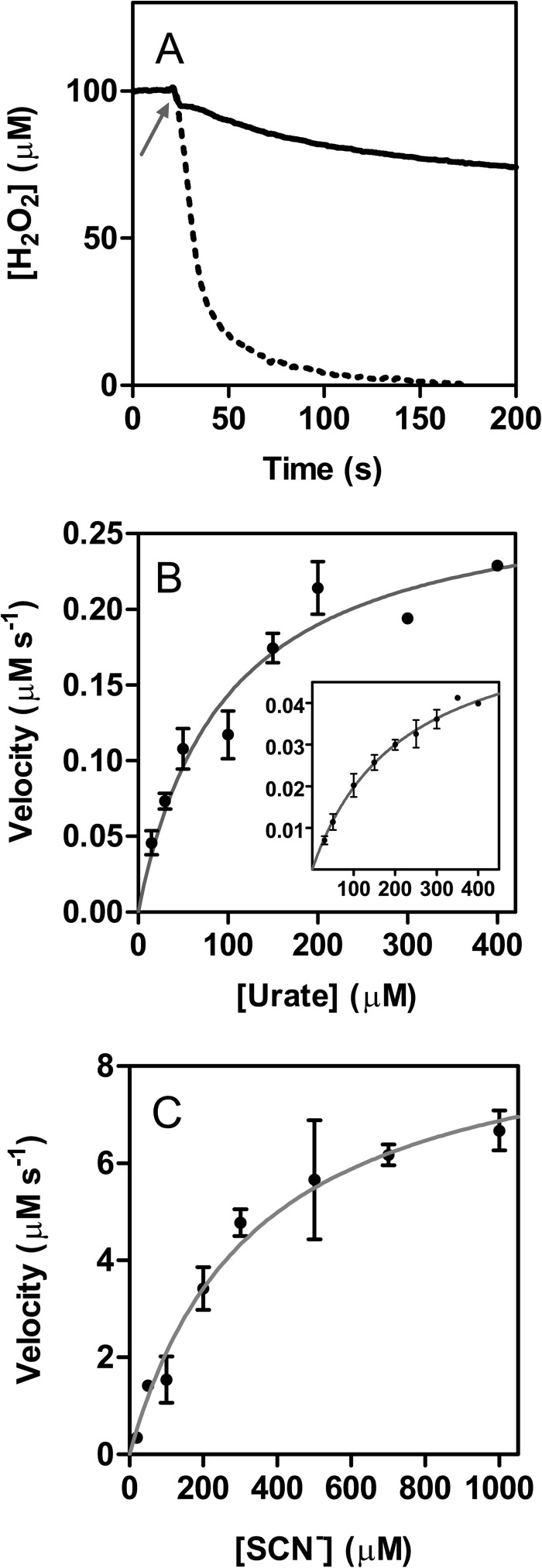
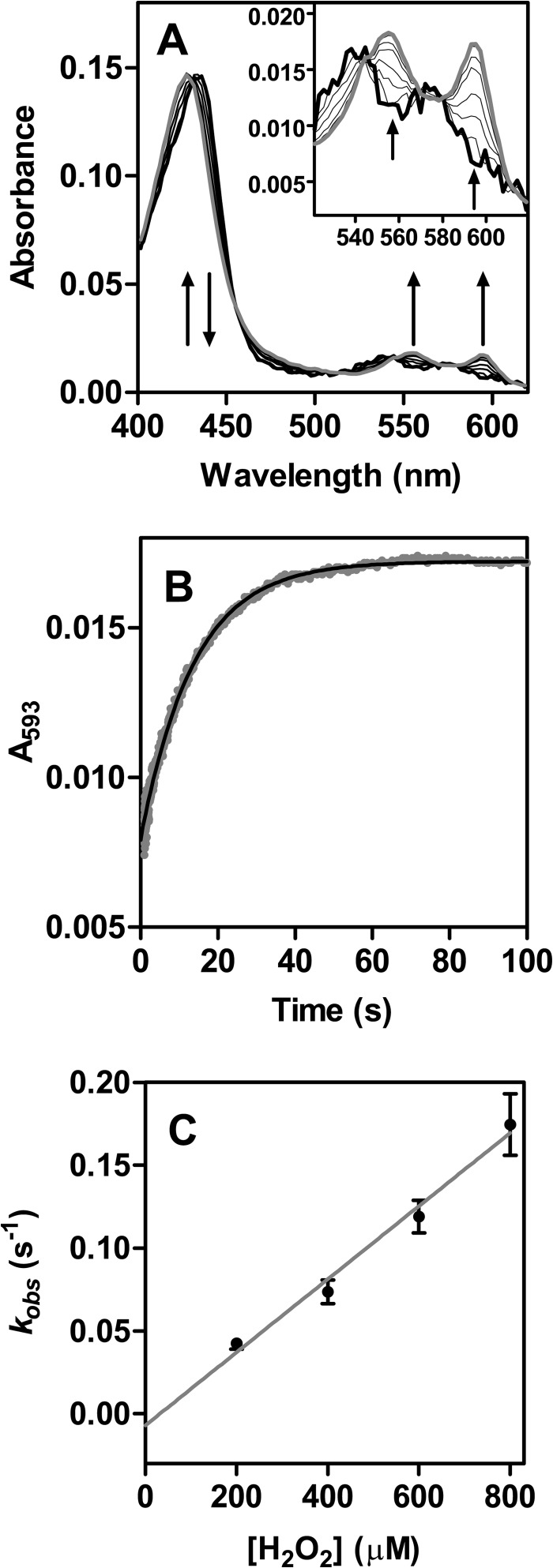
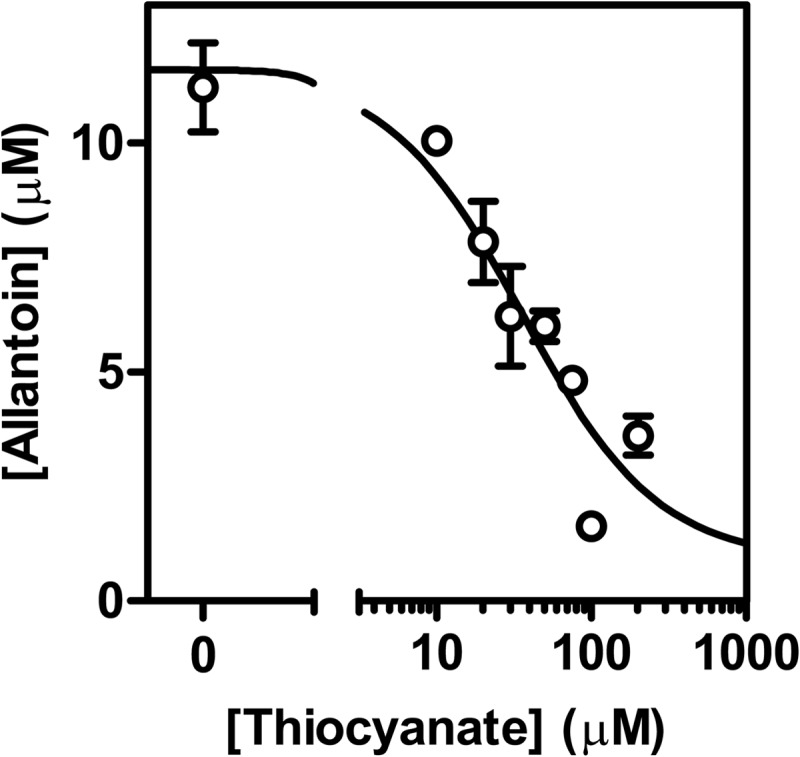
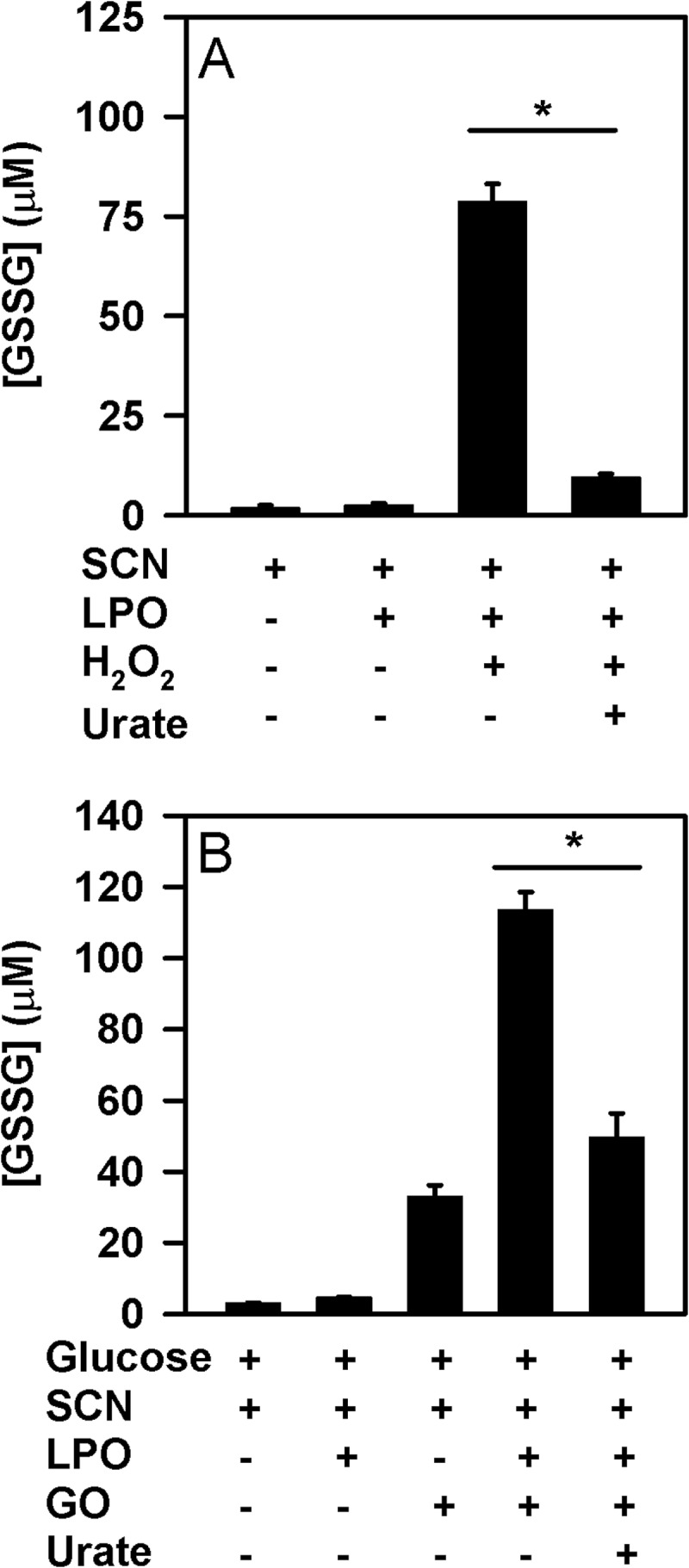
The physiological function of urate is poorly understood. It may act as a danger signal, an antioxidant, or a substrate for heme peroxidases. Whether it reacts sufficiently rapidly with lactoperoxidase (LPO) to act as a physiological substrate remains unknown. LPO is a mammalian peroxidase that plays a key role in the innate immune defense by oxidizing thiocyanate to the bactericidal and fungicidal agent hypothiocyanite. We now demonstrate that urate is a good substrate for bovine LPO. Urate was oxidized by LPO to produce the electrophilic intermediates dehydrourate and 5-hydroxyisourate, which decayed to allantoin. In the presence of superoxide, high yields of hydroperoxides were formed by LPO and urate. Using stopped-flow spectroscopy, we determined rate constants for the reaction of urate with compound I (k1 = 1.1 × 10(7) M(-1) s(-1)) and compound II (k2 = 8.5 × 10(3) M(-1) s(-1)). During urate oxidation, LPO was diverted from its peroxidase cycle because hydrogen peroxide reacted with compound II to give compound III. At physiologically relevant concentrations, urate competed effectively with thiocyanate, the main substrate of LPO for oxidation, and inhibited production of hypothiocyanite. Similarly, hypothiocyanite-dependent killing of Pseudomonas aeruginosa was inhibited by urate. Allantoin was present in human saliva and associated with the concentration of LPO. When hydrogen peroxide was added to saliva, oxidation of urate was dependent on its concentration and peroxidase activity. Our findings establish urate as a likely physiological substrate for LPO that will influence host defense and give rise to reactive electrophilic metabolites.
Keywords: Host Defense; Kinetics; Peroxidase; Pre-steady-state Kinetics; Uric Acid.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Becker B. F. (1993) Towards the physiological function of uric acid. Free Radic. Biol. Med. 14, 615–631 - PubMed
-
- Wayner D. D. M., Burton G. W., Ingold K. U., Barclay L. R. C., Locke S. J. (1987) The relative contributions of vitamin E, urate, ascorbate and proteins to the total peroxyl radical-trapping antioxidant activity of human blood plasma. Biochim. Biophys. Acta 924, 408–419 - PubMed
-
- Uotila J., Metsa-Ketela T., Tuimala R. (1992) Plasma peroxyl radical-trapping capacity in severe preeclampsia is strongly related to uric acid. Clin. Exp. Hypertens. B b11, 71–80
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
