

Biochemical and clinical features of hereditary hyperprolinemia
- PMID: 24931297
- PMCID: PMC4282441
- DOI: 10.1111/ped.12420
Biochemical and clinical features of hereditary hyperprolinemia
Abstract
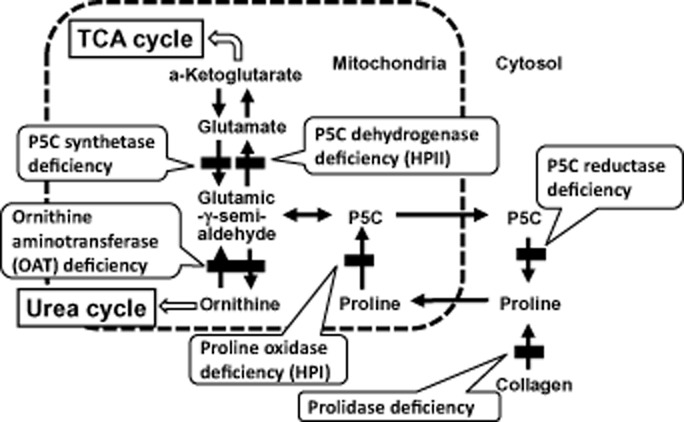
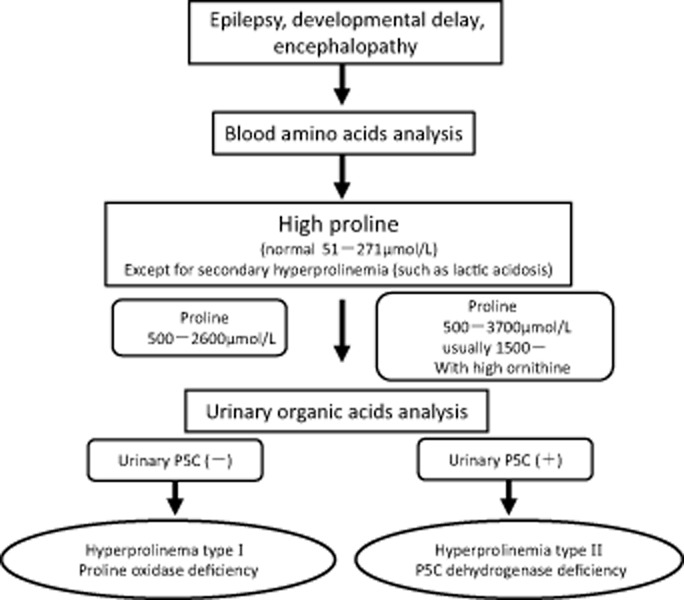
There are two classifications of hereditary hyperprolinemia: type I (HPI) and type II (HPII). Each type is caused by an autosomal recessive inborn error of the proline metabolic pathway. HPI is caused by an abnormality in the proline-oxidizing enzyme (POX). HPII is caused by a deficiency of Δ-1-pyrroline-5-carboxylate (P5C) dehydrogenase (P5CDh). The clinical features of HPI are unclear. Nephropathy, uncontrolled seizures, mental retardation or schizophrenia have been reported in HPI, but a benign phenotype without neurological problems has also been reported. The clinical features of HPII are also unclear. In addition, the precise incidences of HPI and HPII are unknown. Only two cases of HPI and one case of HPII have been identified in Japan through a questionnaire survey and by a study of previous reports. This suggests that hyperprolinemia is a very rare disease in Japan, consistent with earlier reports in Western countries. The one case of HPII found in Japan was diagnosed in an individual with influenza-associated encephalopathy. This suggests that HPII might reduce the threshold for convulsions, thereby increasing the sensitivity of individuals with influenza-associated encephalopathy. The current study presents diagnostic criteria for HPI and HPII, based on plasma proline level, with or without measurements of urinary P5C. In the future, screening for HPI and HPII in healthy individuals, or patients with relatively common diseases such as developmental disabilities, epilepsy, schizophrenia or behavioral problems will be important.
Keywords: P5C; hyperprolinemia type I; hyperprolinemia type II; inborn error of metabolism; proline.
© 2014 The Authors. Pediatrics International published by Wiley Publishing Asia Pty Ltd on behalf of Japan Pediatric Society.
Figures
References
-
- Phang JM, Hu C-A, Valle D. Disorders of proline and hydroxyproline metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 1821–1838. 8th edn.
-
- Mitsubuchi H, Nakamura K, Matsumoto S, Endo F. Inborn errors of proline metabolism. J. Nutr. 2008;138:2016S–2020. - PubMed
-
- Schafer IA, Scriver CR, Efron ML. Familial hyperprolinemia, cerebral dysfunction and renal anomalies occurring in a family with hereditary nephropathy and deafness. N. Engl. J. Med. 1962;267:51–60. - PubMed
-
- Kopelman H, Asatoor AM, Milne MD. Hyperprolinaemia and hereditary nephritis. Lancet. 1964;13:1075–1079. - PubMed
-
- Efron ML. Familial hyperprolinemia. Report of a second case, associated with congenital renal malformations, hereditary hematuria and mild mental retardation, with demonstration of an enzyme defect. N. Engl. J. Med. 1965;272:1243–1254. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
