

Accurate viral population assembly from ultra-deep sequencing data
- PMID: 24932001
- PMCID: PMC4058922
- DOI: 10.1093/bioinformatics/btu295
Accurate viral population assembly from ultra-deep sequencing data
Abstract
Motivation: Next-generation sequencing technologies sequence viruses with ultra-deep coverage, thus promising to revolutionize our understanding of the underlying diversity of viral populations. While the sequencing coverage is high enough that even rare viral variants are sequenced, the presence of sequencing errors makes it difficult to distinguish between rare variants and sequencing errors.
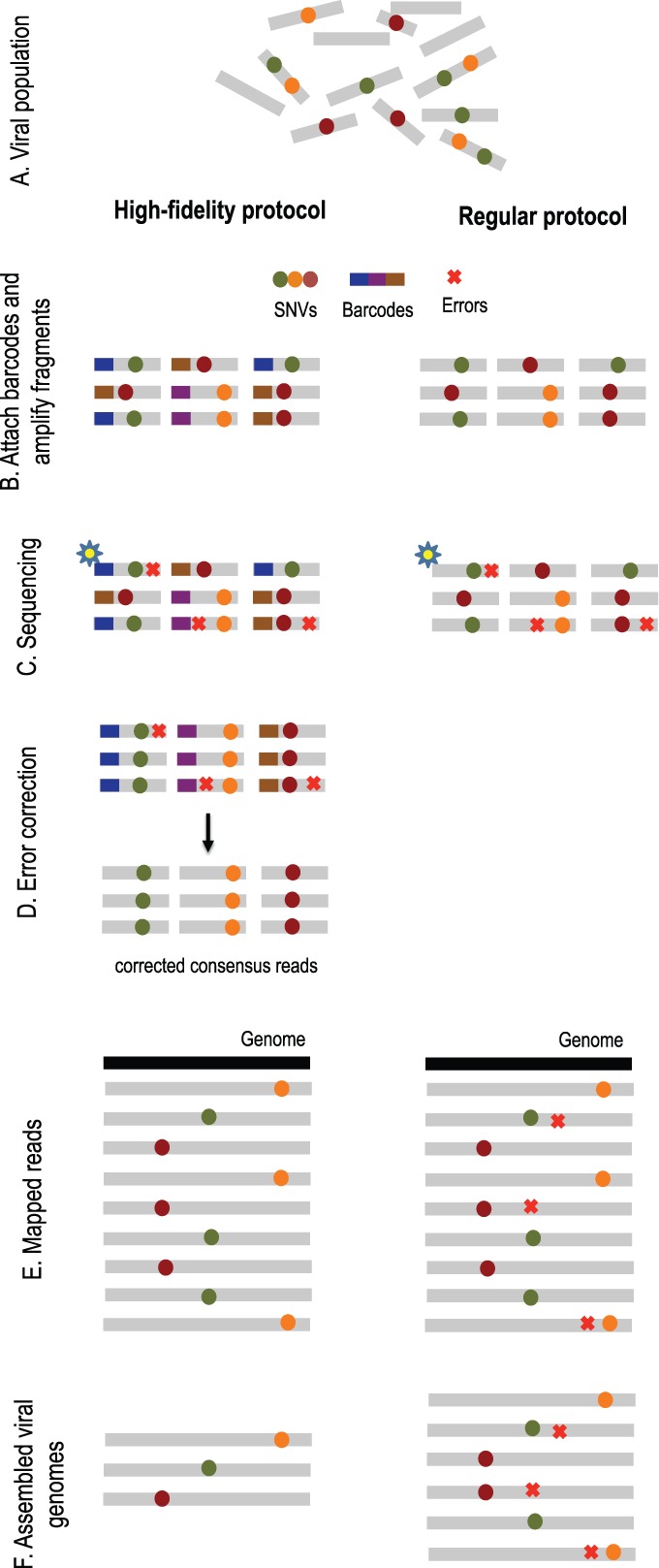
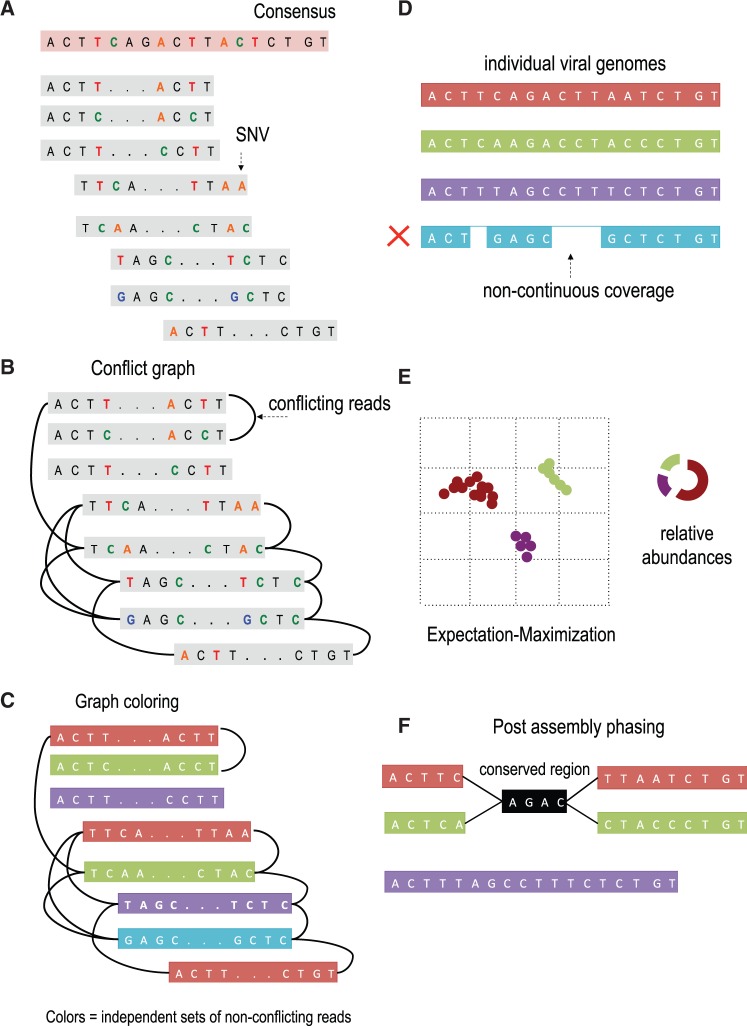
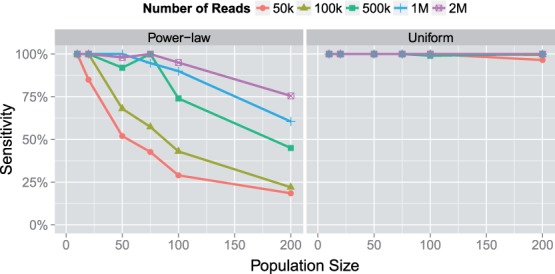
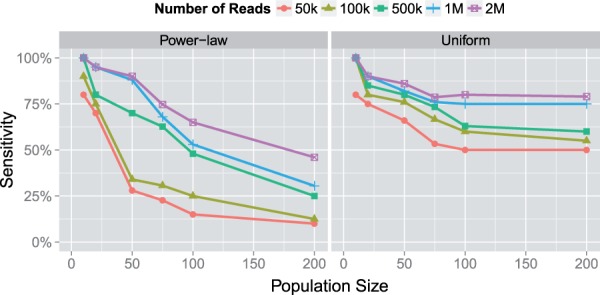
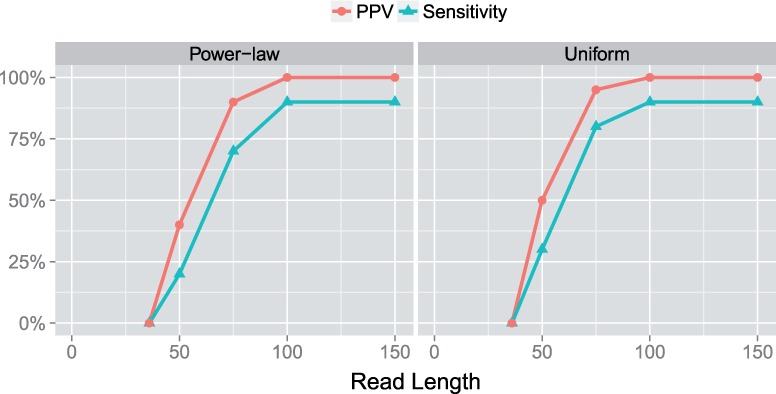
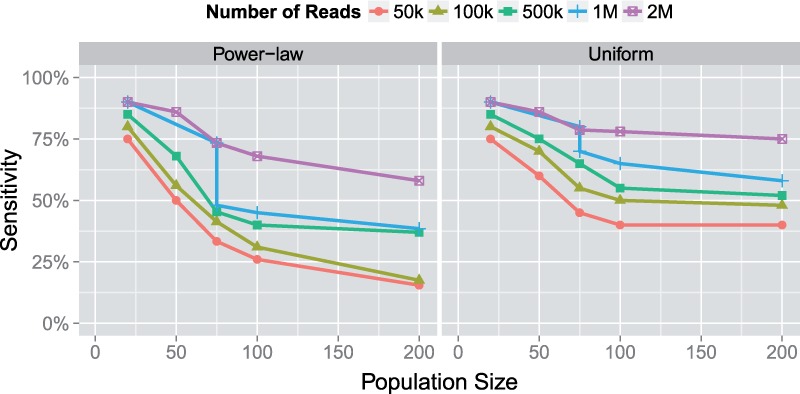
Results: In this article, we present a method to overcome the limitations of sequencing technologies and assemble a diverse viral population that allows for the detection of previously undiscovered rare variants. The proposed method consists of a high-fidelity sequencing protocol and an accurate viral population assembly method, referred to as Viral Genome Assembler (VGA). The proposed protocol is able to eliminate sequencing errors by using individual barcodes attached to the sequencing fragments. Highly accurate data in combination with deep coverage allow VGA to assemble rare variants. VGA uses an expectation-maximization algorithm to estimate abundances of the assembled viral variants in the population. RESULTS on both synthetic and real datasets show that our method is able to accurately assemble an HIV viral population and detect rare variants previously undetectable due to sequencing errors. VGA outperforms state-of-the-art methods for genome-wide viral assembly. Furthermore, our method is the first viral assembly method that scales to millions of sequencing reads.
Availability: Our tool VGA is freely available at http://genetics.cs.ucla.edu/vga/
© The Author 2014. Published by Oxford University Press.
Figures
References
-
- Armin, T, Beerenwinkel N. 2013 http://www.bsse.ethz.ch/cbg/software/InDelFixer.
-
- Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics. 2008;24:i153–i159. - PubMed
Publication types
MeSH terms
Grants and funding
- R01-GM083198/GM/NIGMS NIH HHS/United States
- R01 MH101782/MH/NIMH NIH HHS/United States
- P01- HL30568/HL/NHLBI NIH HHS/United States
- U01-DA024417/DA/NIDA NIH HHS/United States
- K25 HL080079/HL/NHLBI NIH HHS/United States
- P01 HL028481/HL/NHLBI NIH HHS/United States
- U01 DA024417/DA/NIDA NIH HHS/United States
- R01-MH101782/MH/NIMH NIH HHS/United States
- K25-HL080079/HL/NHLBI NIH HHS/United States
- R01 ES022282/ES/NIEHS NIH HHS/United States
- P01 HL030568/HL/NHLBI NIH HHS/United States
- R01 GM083198/GM/NIGMS NIH HHS/United States
- R01-ES022282/ES/NIEHS NIH HHS/United States
- P01-HL28481/HL/NHLBI NIH HHS/United States
- P30 CA016042/CA/NCI NIH HHS/United States
- P30 AI028697/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
