

Phenotype prediction based on genome-wide DNA methylation data
- PMID: 24934728
- PMCID: PMC4073816
- DOI: 10.1186/1471-2105-15-193
Phenotype prediction based on genome-wide DNA methylation data
Abstract
Background: DNA methylation (DNAm) has important regulatory roles in many biological processes and diseases. It is the only epigenetic mark with a clear mechanism of mitotic inheritance and the only one easily available on a genome scale. Aberrant cytosine-phosphate-guanine (CpG) methylation has been discussed in the context of disease aetiology, especially cancer. CpG hypermethylation of promoter regions is often associated with silencing of tumour suppressor genes and hypomethylation with activation of oncogenes.Supervised principal component analysis (SPCA) is a popular machine learning method. However, in a recent application to phenotype prediction from DNAm data SPCA was inferior to the specific method EVORA.
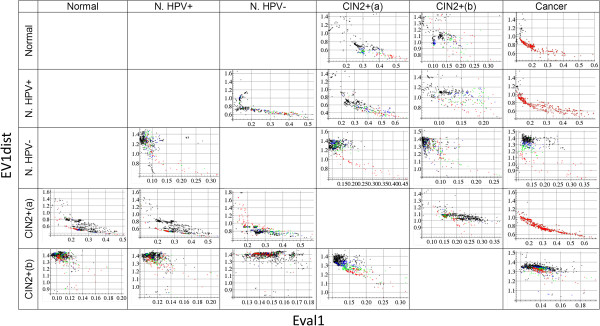
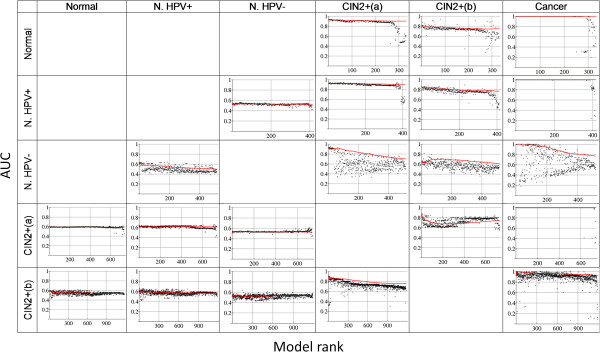
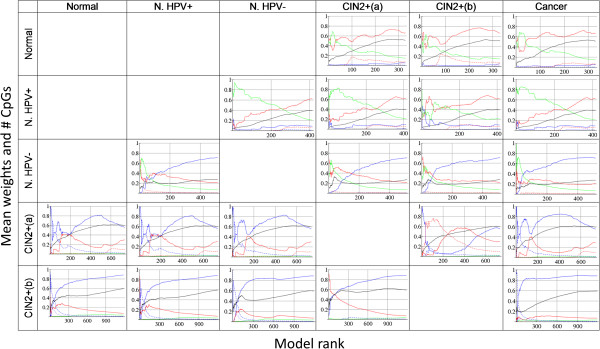
Results: We present Model-Selection-SPCA (MS-SPCA), an enhanced version of SPCA. MS-SPCA applies several models that perform well in the training data to the test data and selects the very best models for final prediction based on parameters of the test data.We have applied MS-SPCA for phenotype prediction from genome-wide DNAm data. CpGs used for prediction are selected based on the quantification of three features of their methylation (average methylation difference, methylation variation difference and methylation-age-correlation). We analysed four independent case-control datasets that correspond to different stages of cervical cancer: (i) cases currently cytologically normal, but will later develop neoplastic transformations, (ii, iii) cases showing neoplastic transformations and (iv) cases with confirmed cancer. The first dataset was split into several smaller case-control datasets (samples either Human Papilloma Virus (HPV) positive or negative). We demonstrate that cytology normal HPV+ and HPV- samples contain DNAm patterns which are associated with later neoplastic transformations. We present evidence that DNAm patterns exist in cytology normal HPV- samples that (i) predispose to neoplastic transformations after HPV infection and (ii) predispose to HPV infection itself. MS-SPCA performs significantly better than EVORA.
Conclusions: MS-SPCA can be applied to many classification problems. Additional improvements could include usage of more than one principal component (PC), with automatic selection of the optimal number of PCs. We expect that MS-SPCA will be useful for analysing recent larger DNAm data to predict future neoplastic transformations.
Figures
References
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
-
- Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13:705–719. - PubMed
-
- McKay JA, Mathers JC. Diet induced epigenetic changes and their implications for health. Acta Physiol (Oxf) 2011;202:103–118. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
