

A new approach for efficient genotype imputation using information from relatives
- PMID: 24935670
- PMCID: PMC4076979
- DOI: 10.1186/1471-2164-15-478
A new approach for efficient genotype imputation using information from relatives
Abstract
Background: Genotype imputation can help reduce genotyping costs particularly for implementation of genomic selection. In applications entailing large populations, recovering the genotypes of untyped loci using information from reference individuals that were genotyped with a higher density panel is computationally challenging. Popular imputation methods are based upon the Hidden Markov model and have computational constraints due to an intensive sampling process. A fast, deterministic approach, which makes use of both family and population information, is presented here. All individuals are related and, therefore, share haplotypes which may differ in length and frequency based on their relationships. The method starts with family imputation if pedigree information is available, and then exploits close relationships by searching for long haplotype matches in the reference group using overlapping sliding windows. The search continues as the window size is shrunk in each chromosome sweep in order to capture more distant relationships.
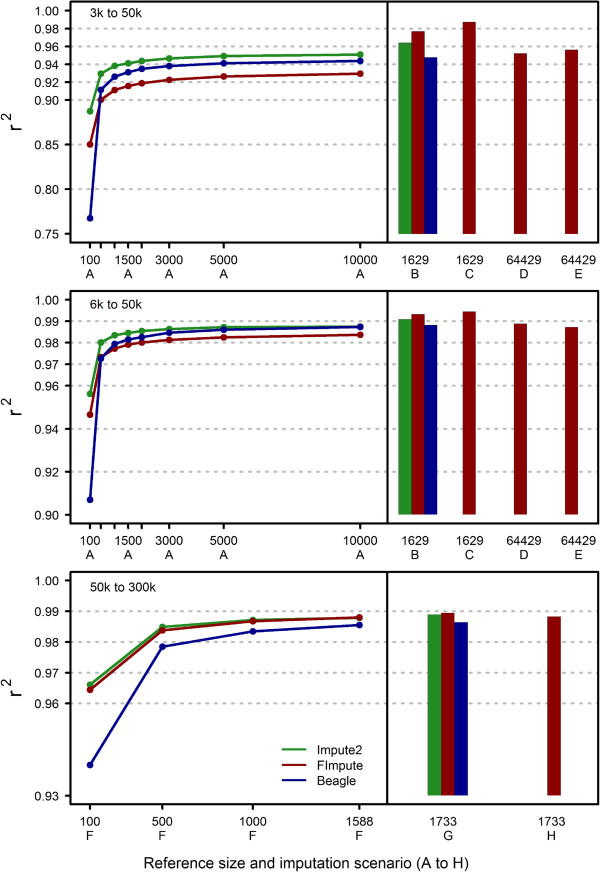
Results: The proposed method gave higher or similar imputation accuracy than Beagle and Impute2 in cattle data sets when all available information was used. When close relatives of target individuals were present in the reference group, the method resulted in higher accuracy compared to the other two methods even when the pedigree was not used. Rare variants were also imputed with higher accuracy. Finally, computing requirements were considerably lower than those of Beagle and Impute2. The presented method took 28 minutes to impute from 6 k to 50 k genotypes for 2,000 individuals with a reference size of 64,429 individuals.
Conclusions: The proposed method efficiently makes use of information from close and distant relatives for accurate genotype imputation. In addition to its high imputation accuracy, the method is fast, owing to its deterministic nature and, therefore, it can easily be used in large data sets where the use of other methods is impractical.
Figures


References
-
- Nejati-Javaremi A, Smith C, Gibson JP. Effect of total allelic relationship on accuracy of evaluation and response to selection. J Anim Sci. 1997;75:1738–1745. - PubMed
-
- Van der Werf JHJ. Potential benefit of genomic selection in sheep. Proc Assoc Advanc Anim Genetics. 2009;18:38–41.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
