

Pharmacologic management of Duchenne muscular dystrophy: target identification and preclinical trials
- PMID: 24936034
- PMCID: PMC4158345
- DOI: 10.1093/ilar/ilu011
Pharmacologic management of Duchenne muscular dystrophy: target identification and preclinical trials
Abstract
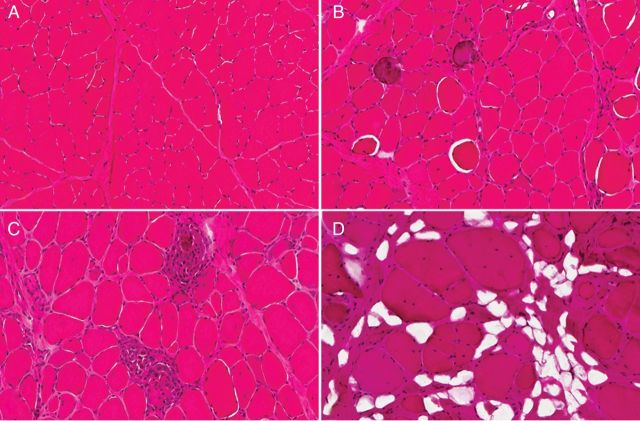
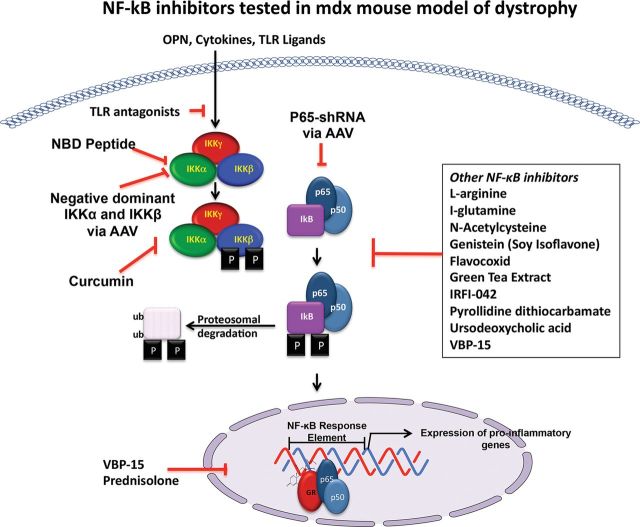
Duchenne muscular dystrophy (DMD) is an X-linked human disorder in which absence of the protein dystrophin causes degeneration of skeletal and cardiac muscle. For the sake of treatment development, over and above definitive genetic and cell-based therapies, there is considerable interest in drugs that target downstream disease mechanisms. Drug candidates have typically been chosen based on the nature of pathologic lesions and presumed underlying mechanisms and then tested in animal models. Mammalian dystrophinopathies have been characterized in mice (mdx mouse) and dogs (golden retriever muscular dystrophy [GRMD]). Despite promising results in the mdx mouse, some therapies have not shown efficacy in DMD. Although the GRMD model offers a higher hurdle for translation, dogs have primarily been used to test genetic and cellular therapies where there is greater risk. Failed translation of animal studies to DMD raises questions about the propriety of methods and models used to identify drug targets and test efficacy of pharmacologic intervention. The mdx mouse and GRMD dog are genetically homologous to DMD but not necessarily analogous. Subcellular species differences are undoubtedly magnified at the whole-body level in clinical trials. This problem is compounded by disparate cultures in clinical trials and preclinical studies, pointing to a need for greater rigor and transparency in animal experiments. Molecular assays such as mRNA arrays and genome-wide association studies allow identification of genetic drug targets more closely tied to disease pathogenesis. Genes in which polymorphisms have been directly linked to DMD disease progression, as with osteopontin, are particularly attractive targets.
Keywords: Duchenne muscular dystrophy; animal models; drug development; genome wide association studies; golden retriever muscular dystrophy; mRNA arrays; mdx mouse; preclinical studies.
© The Author 2014. Published by Oxford University Press on behalf of the Institute for Laboratory Animal Research. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures






References
-
- Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. - PMC - PubMed
-
- Allen DG, Gervasio OL, Yeung EW, Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol. 2010;88:83–91. - PubMed
-
- Ambrósio CE, Valadares MC, Zucconi E, Cabral R, Pearson PL, Gaiad TP, Canovas M, Vainzof M, Miglino MA, Zatz M. Ringo, a Golden Retriever Muscular Dystrophy (GRMD) dog with absent dystrophin but normal strength. Neuromuscul Disord. 2008;18:892–893. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 AR050478/AR/NIAMS NIH HHS/United States
- K12 HD001399/HD/NICHD NIH HHS/United States
- 1U24NS059696-01A1/NS/NINDS NIH HHS/United States
- K26OD011171/OD/NIH HHS/United States
- 1U54HD053177-01A1/HD/NICHD NIH HHS/United States
- U54 HD053177/HD/NICHD NIH HHS/United States
- 5R24HD050846/HD/NICHD NIH HHS/United States
- 1F32AR060703-01/AR/NIAMS NIH HHS/United States
- K26 OD011171/OD/NIH HHS/United States
- U24 NS059696/NS/NINDS NIH HHS/United States
- F32 AR060703/AR/NIAMS NIH HHS/United States
- K12HD001399-04/HD/NICHD NIH HHS/United States
- R24 HD050846/HD/NICHD NIH HHS/United States
- R01-AR050478/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
