

Stenotrophomonas comparative genomics reveals genes and functions that differentiate beneficial and pathogenic bacteria
- PMID: 24939220
- PMCID: PMC4101175
- DOI: 10.1186/1471-2164-15-482
Stenotrophomonas comparative genomics reveals genes and functions that differentiate beneficial and pathogenic bacteria
Abstract
Background: In recent years, the number of human infections caused by opportunistic pathogens has increased dramatically. Plant rhizospheres are one of the most typical natural reservoirs for these pathogens but they also represent a great source for beneficial microbes with potential for biotechnological applications. However, understanding the natural variation and possible differences between pathogens and beneficials is the main challenge in furthering these possibilities. The genus Stenotrophomonas contains representatives found to be associated with human and plant host.
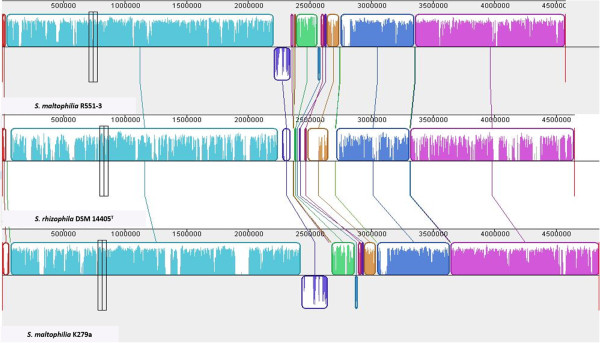
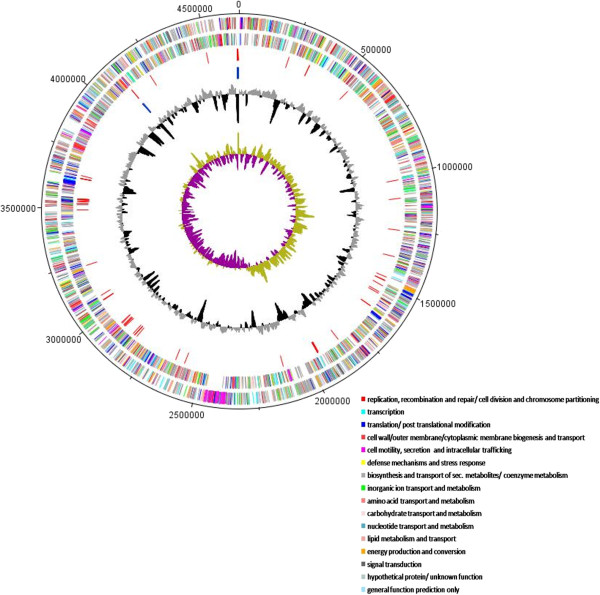
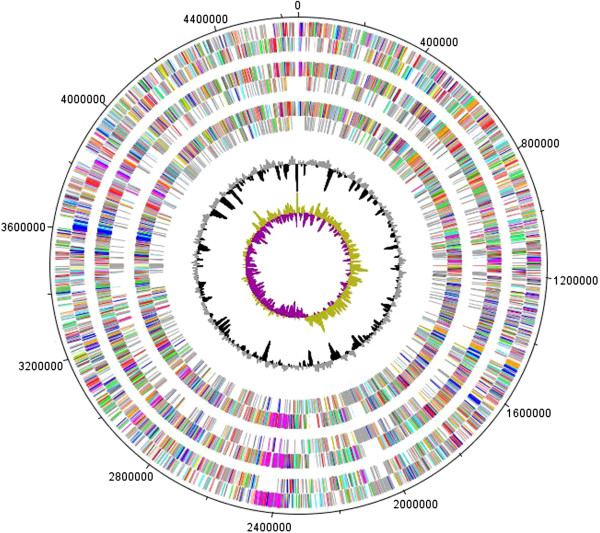
Results: We used comparative genomics as well as transcriptomic and physiological approaches to detect significant borders between the Stenotrophomonas strains: the multi-drug resistant pathogenic S. maltophilia and the plant-associated strains S. maltophilia R551-3 and S. rhizophila DSM14405T (both are biocontrol agents). We found an overall high degree of sequence similarity between the genomes of all three strains. Despite the notable similarity in potential factors responsible for host invasion and antibiotic resistance, other factors including several crucial virulence factors and heat shock proteins were absent in the plant-associated DSM14405T. Instead, S. rhizophila DSM14405T possessed unique genes for the synthesis and transport of the plant-protective spermidine, plant cell-wall degrading enzymes, and high salinity tolerance. Moreover, the presence or absence of bacterial growth at 37°C was identified as a very simple method in differentiating between pathogenic and non-pathogenic isolates. DSM14405T is not able to grow at this human-relevant temperature, most likely in great part due to the absence of heat shock genes and perhaps also because of the up-regulation at increased temperatures of several genes involved in a suicide mechanism.
Conclusions: While this study is important for understanding the mechanisms behind the emerging pattern of infectious diseases, it is, to our knowledge, the first of its kind to assess the risk of beneficial strains for biotechnological applications. We identified certain traits typical of pathogens such as growth at the human body temperature together with the production of heat shock proteins as opposed to a temperature-regulated suicide system that is harnessed by beneficials.
Figures



References
-
- Hartmann A, Rothballer M, Schmid M. Lorenz Hiltner, a pioneer in rhizosphere microbial ecology and soil bacteriology research. Plant and Soil. 2008;312(1–2):7–14. doi: 10.1007/s11104-007-9514-z. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
