

The yeast deletion collection: a decade of functional genomics
- PMID: 24939991
- PMCID: PMC4063906
- DOI: 10.1534/genetics.114.161620
The yeast deletion collection: a decade of functional genomics
Abstract
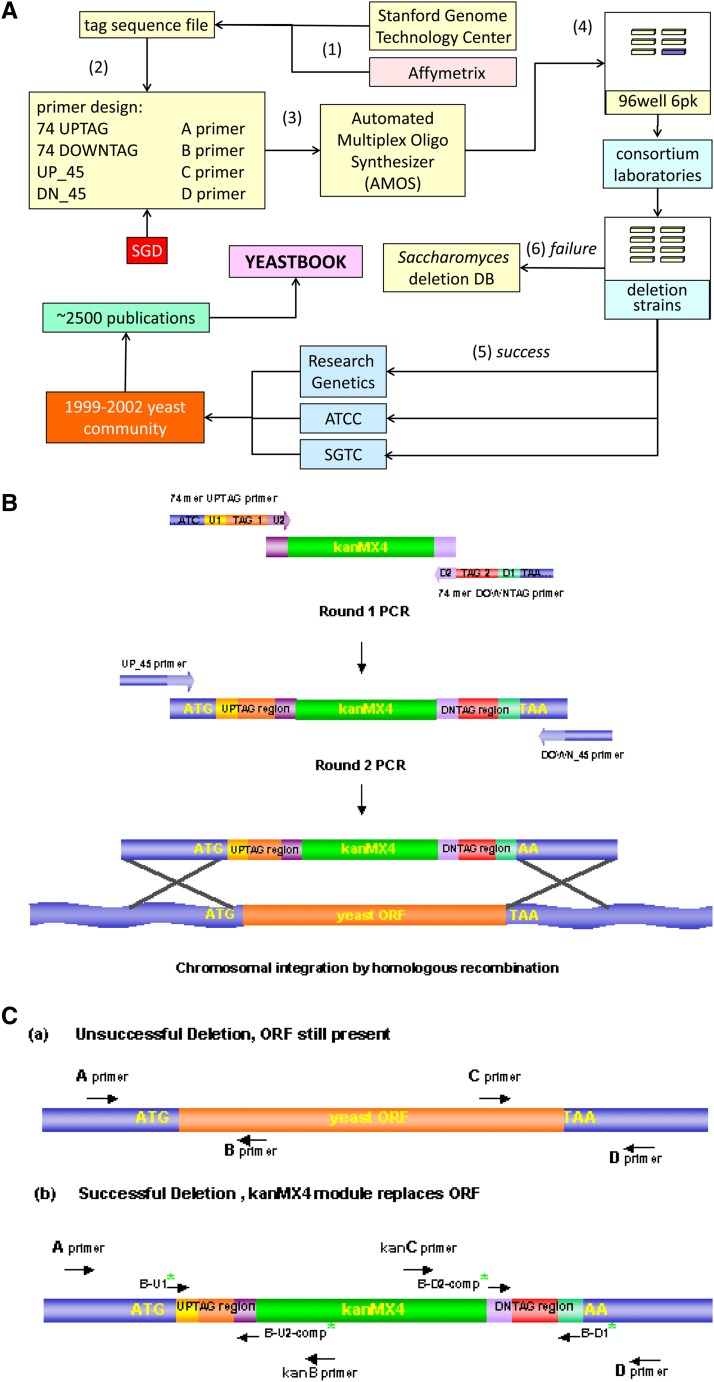
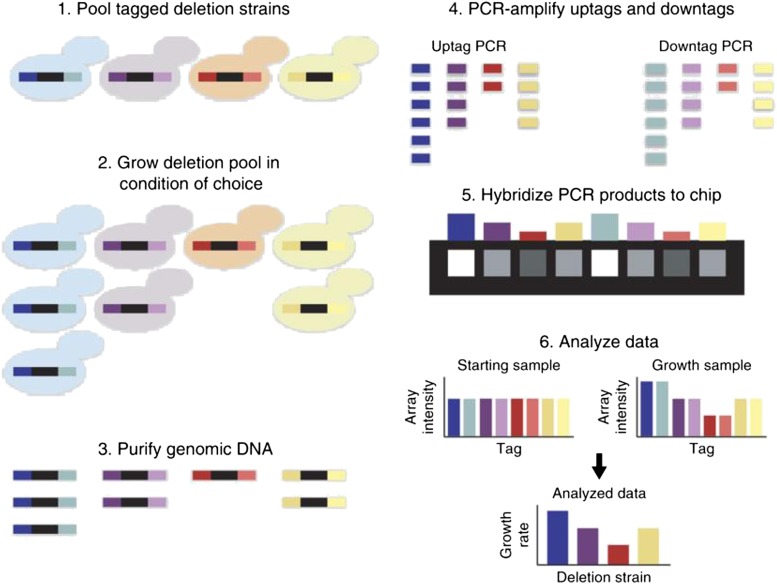
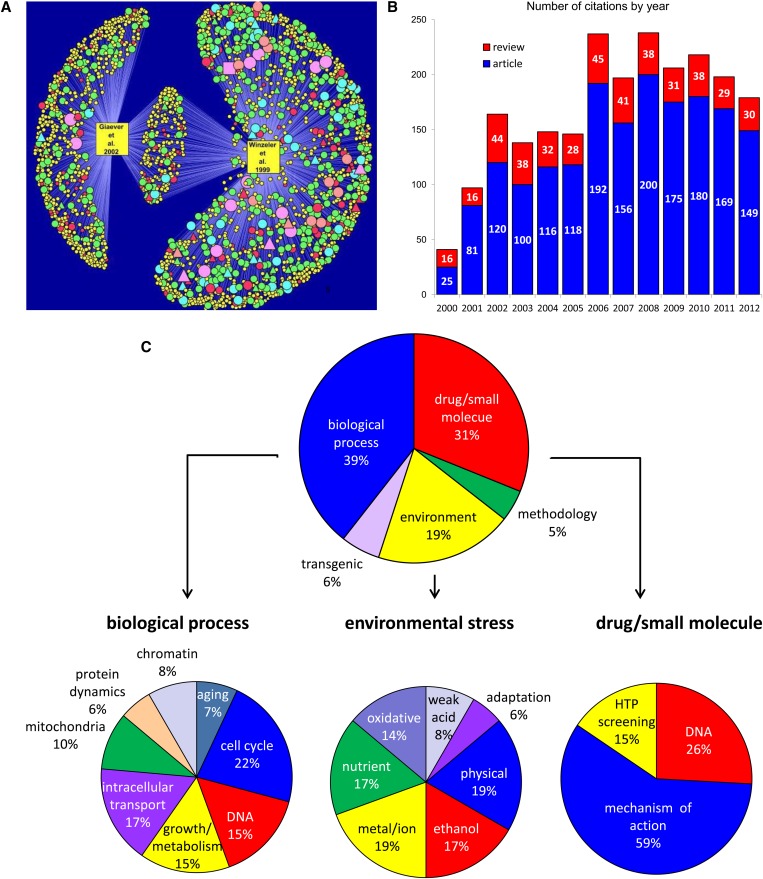
The yeast deletion collections comprise >21,000 mutant strains that carry precise start-to-stop deletions of ∼6000 open reading frames. This collection includes heterozygous and homozygous diploids, and haploids of both MAT A: and MATα mating types. The yeast deletion collection, or yeast knockout (YKO) set, represents the first and only complete, systematically constructed deletion collection available for any organism. Conceived during the Saccharomyces cerevisiae sequencing project, work on the project began in 1998 and was completed in 2002. The YKO strains have been used in numerous laboratories in >1000 genome-wide screens. This landmark genome project has inspired development of numerous genome-wide technologies in organisms from yeast to man. Notable spinoff technologies include synthetic genetic array and HIPHOP chemogenomics. In this retrospective, we briefly describe the yeast deletion project and some of its most noteworthy biological contributions and the impact that these collections have had on the yeast research community and on genomics in general.
Keywords: fitness profiling; functional genomics; mutant phenotype; yeast deletion.
Copyright © 2014 by the Genetics Society of America.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
