

Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ
- PMID: 24942700
- PMCID: PMC4159666
- DOI: 10.1074/mcp.M113.031591
Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ
Abstract
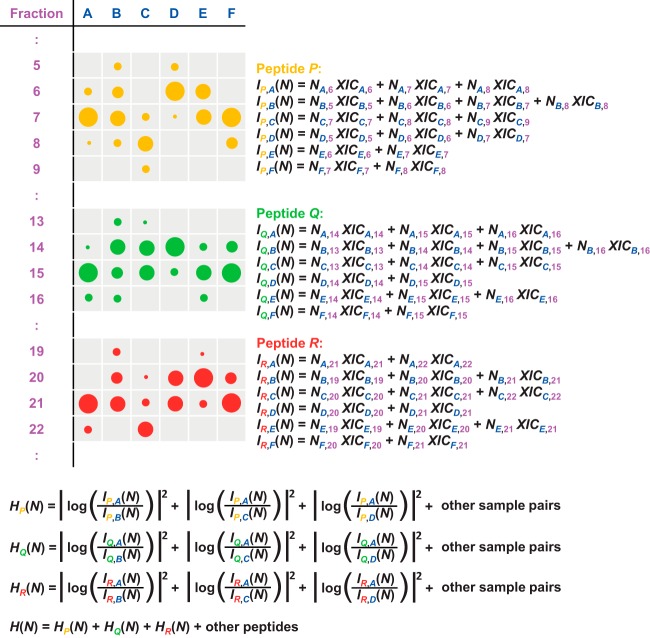
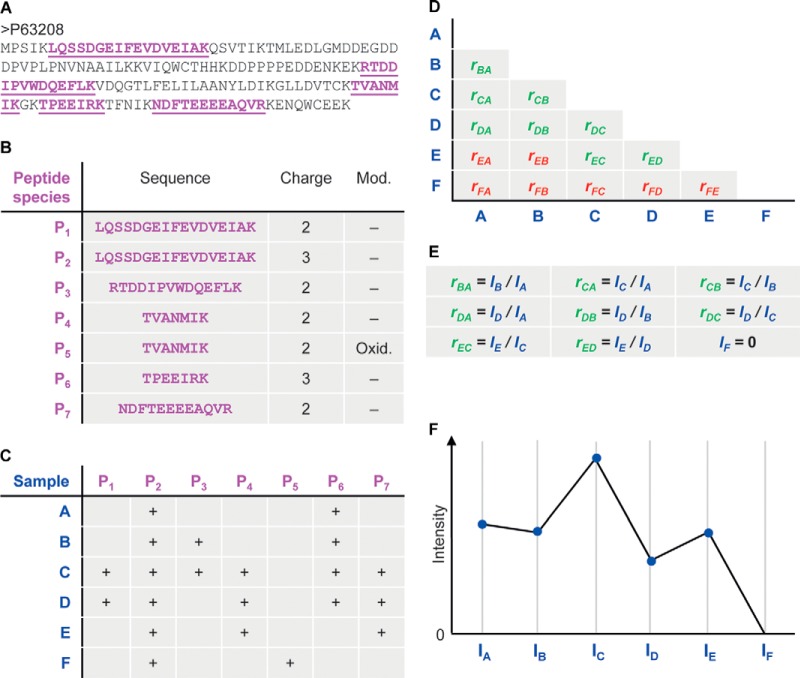
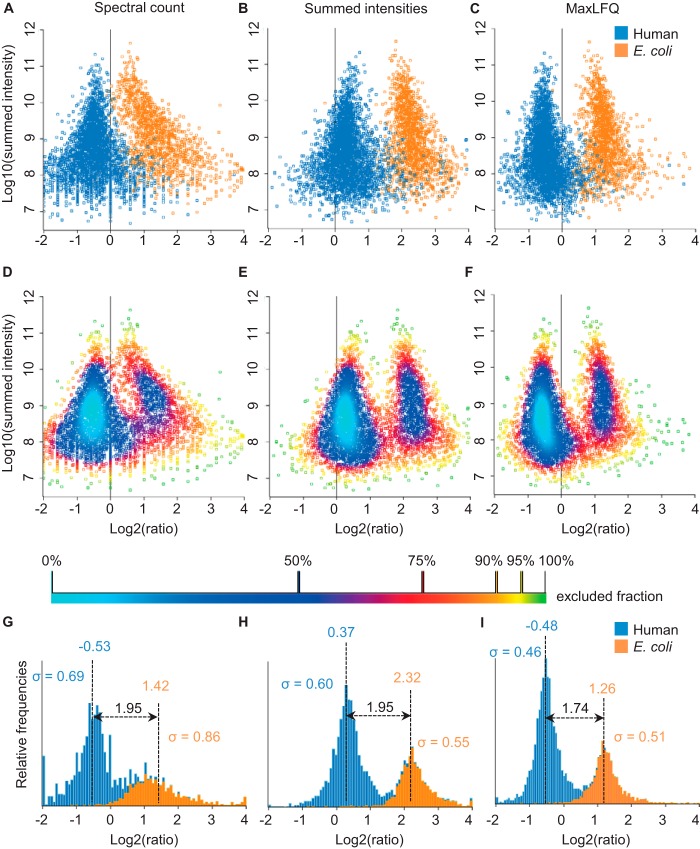
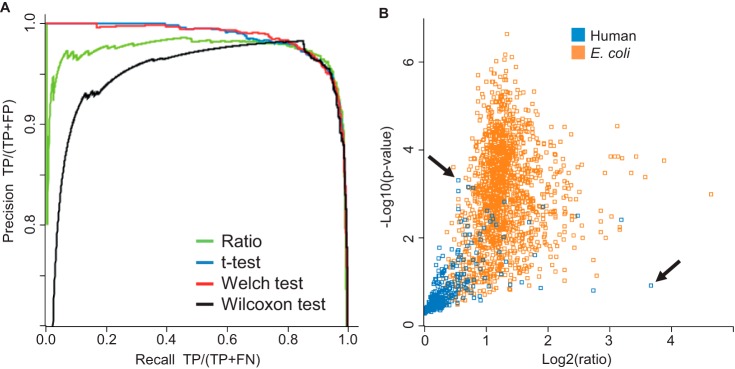
Protein quantification without isotopic labels has been a long-standing interest in the proteomics field. However, accurate and robust proteome-wide quantification with label-free approaches remains a challenge. We developed a new intensity determination and normalization procedure called MaxLFQ that is fully compatible with any peptide or protein separation prior to LC-MS analysis. Protein abundance profiles are assembled using the maximum possible information from MS signals, given that the presence of quantifiable peptides varies from sample to sample. For a benchmark dataset with two proteomes mixed at known ratios, we accurately detected the mixing ratio over the entire protein expression range, with greater precision for abundant proteins. The significance of individual label-free quantifications was obtained via a t test approach. For a second benchmark dataset, we accurately quantify fold changes over several orders of magnitude, a task that is challenging with label-based methods. MaxLFQ is a generic label-free quantification technology that is readily applicable to many biological questions; it is compatible with standard statistical analysis workflows, and it has been validated in many and diverse biological projects. Our algorithms can handle very large experiments of 500+ samples in a manageable computing time. It is implemented in the freely available MaxQuant computational proteomics platform and works completely seamlessly at the click of a button.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
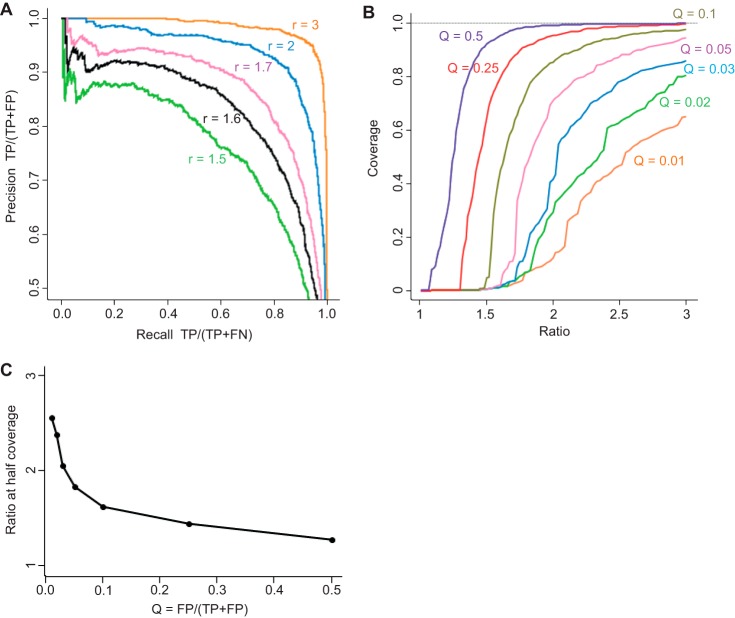
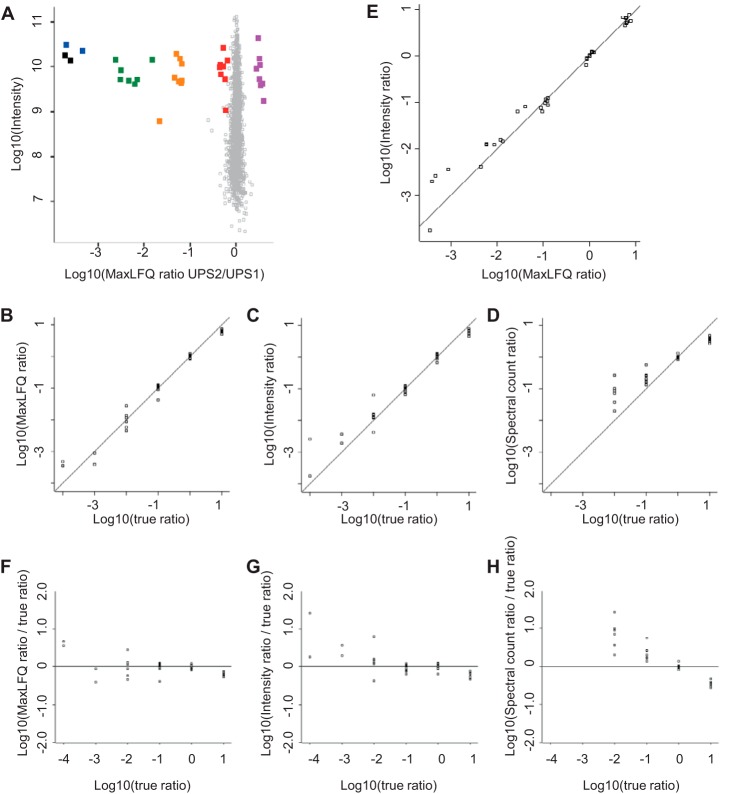
References
-
- Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 - PubMed
-
- Ong S. E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 - PubMed
-
- Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 - PubMed
-
- Cox J., Mann M. (2007) Is proteomics the new genomics? Cell 130, 395–398 - PubMed
-
- Altelaar A. F., Munoz J., Heck A. J. (2013) Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14, 35–48 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
