

Selective targeting of TGF-β activation to treat fibroinflammatory airway disease
- PMID: 24944194
- PMCID: PMC4341974
- DOI: 10.1126/scitranslmed.3008074
Selective targeting of TGF-β activation to treat fibroinflammatory airway disease
Abstract
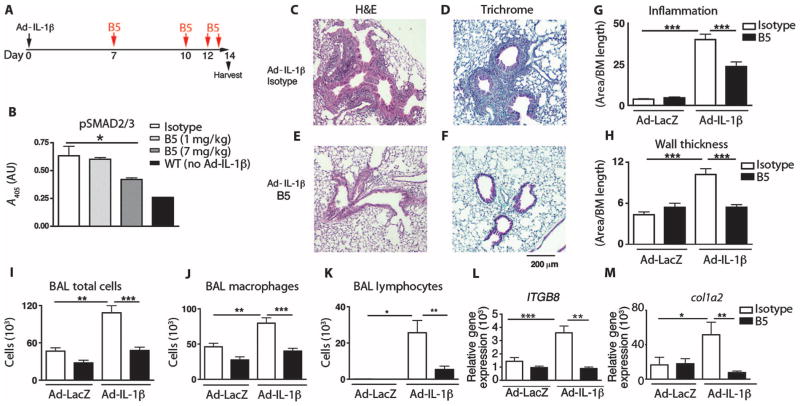
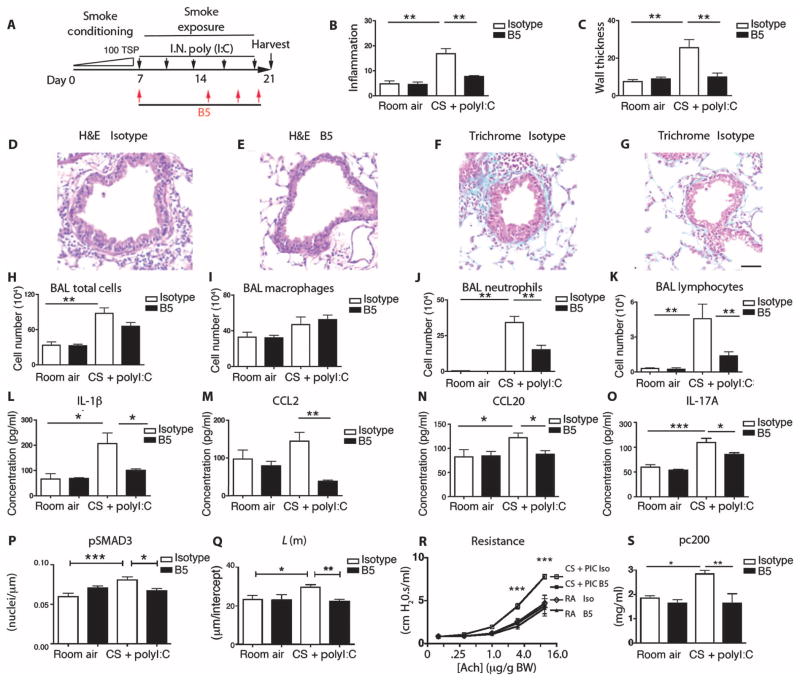
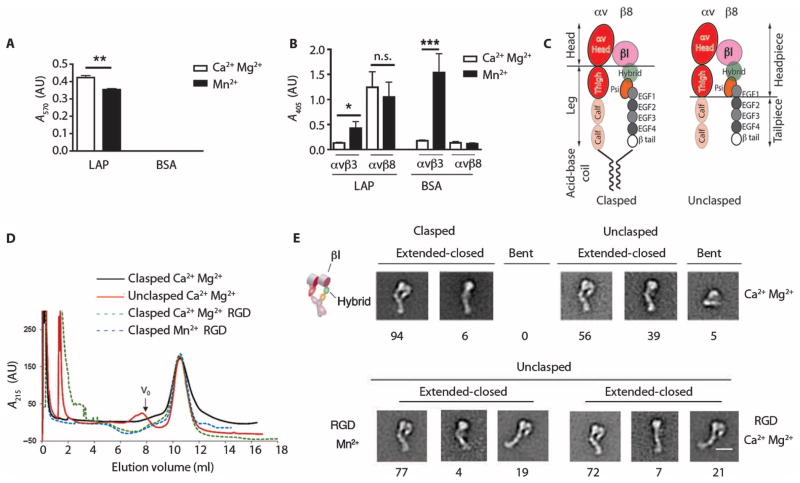
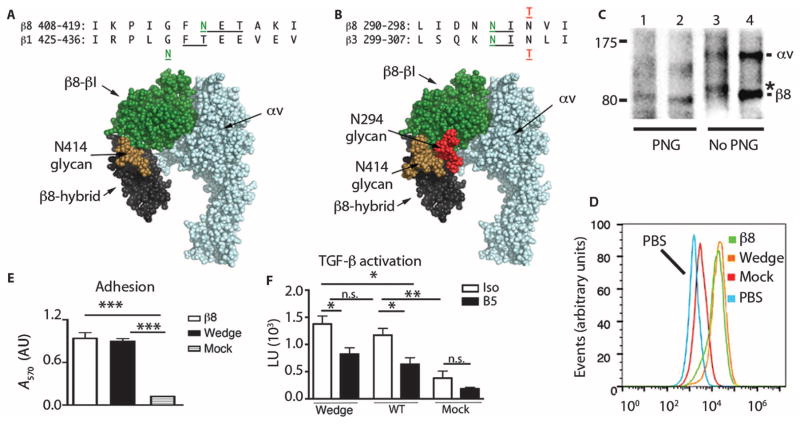
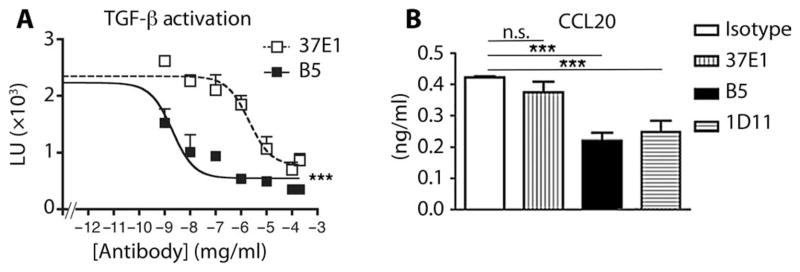
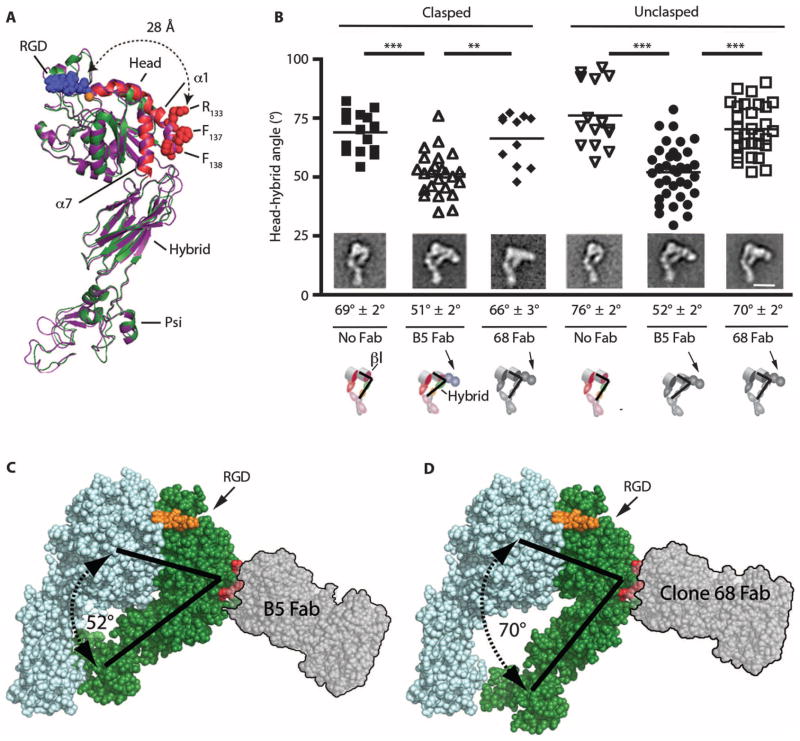
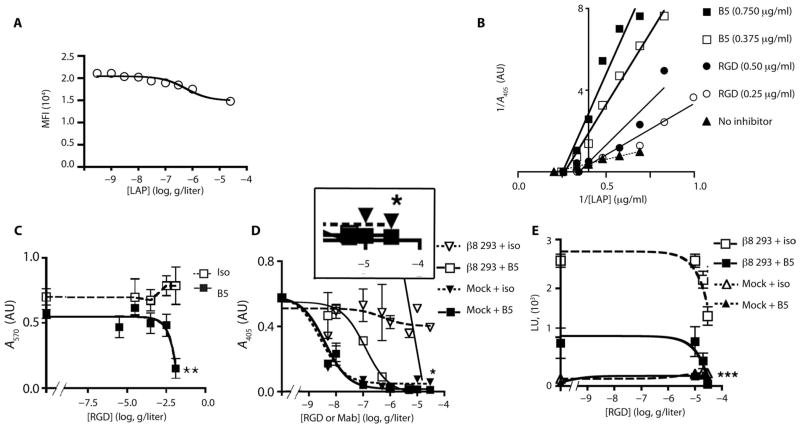
Airway remodeling, caused by inflammation and fibrosis, is a major component of chronic obstructive pulmonary disease (COPD) and currently has no effective treatment. Transforming growth factor-β (TGF-β) has been widely implicated in the pathogenesis of airway remodeling in COPD. TGF-β is expressed in a latent form that requires activation. The integrin αvβ8 (encoded by the itgb8 gene) is a receptor for latent TGF-β and is essential for its activation. Expression of integrin αvβ8 is increased in airway fibroblasts in COPD and thus is an attractive therapeutic target for the treatment of airway remodeling in COPD. We demonstrate that an engineered optimized antibody to human αvβ8 (B5) inhibited TGF-β activation in transgenic mice expressing only human and not mouse ITGB8. The B5 engineered antibody blocked fibroinflammatory responses induced by tobacco smoke, cytokines, and allergens by inhibiting TGF-β activation. To clarify the mechanism of action of B5, we used hydrodynamic, mutational, and electron microscopic methods to demonstrate that αvβ8 predominantly adopts a constitutively active, extended-closed headpiece conformation. Epitope mapping and functional characterization of B5 revealed an allosteric mechanism of action due to locking-in of a low-affinity αvβ8 conformation. Collectively, these data demonstrate a new model for integrin function and present a strategy to selectively target the TGF-β pathway to treat fibroinflammatory airway diseases.
Copyright © 2014, American Association for the Advancement of Science.
Conflict of interest statement
Figures
References
-
- Centers for Disease Control and Prevention (CDC) Chronic obstructive pulmonary disease among adults—United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:938–943. - PubMed
-
- Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:434–439. - PubMed
-
- Han MK, Kazerooni EA, Lynch DA, Liu LX, Murray S, Curtis JL, Criner GJ, Kim V, Bowler RP, Hanania NA, Anzueto AR, Make BJ, Hokanson JE, Crapo JD, Silverman EK, Martinez FJ, Washko GR COPDGene Investigators. Chronic obstructive pulmonary disease exacerbations in the COPDGene study: Associated radiologic phenotypes. Radiology. 2011;261:274–282. - PMC - PubMed
-
- Wilkinson TM, Donaldson GC, Johnston SL, Openshaw PJ, Wedzicha JA. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:871–876. - PubMed
-
- Uhl EW, Castleman WL, Sorkness RL, Busse WW, Lemanske RF, Jr, McAllister PK. Parainfluenza virus-induced persistence of airway inflammation, fibrosis, and dysfunction associated with TGF-beta 1 expression in brown Norway rats. Am J Respir Crit Care Med. 1996;154:1834–1842. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
