

Choice of transcripts and software has a large effect on variant annotation
- PMID: 24944579
- PMCID: PMC4062061
- DOI: 10.1186/gm543
Choice of transcripts and software has a large effect on variant annotation
Abstract
Background: Variant annotation is a crucial step in the analysis of genome sequencing data. Functional annotation results can have a strong influence on the ultimate conclusions of disease studies. Incorrect or incomplete annotations can cause researchers both to overlook potentially disease-relevant DNA variants and to dilute interesting variants in a pool of false positives. Researchers are aware of these issues in general, but the extent of the dependency of final results on the choice of transcripts and software used for annotation has not been quantified in detail.
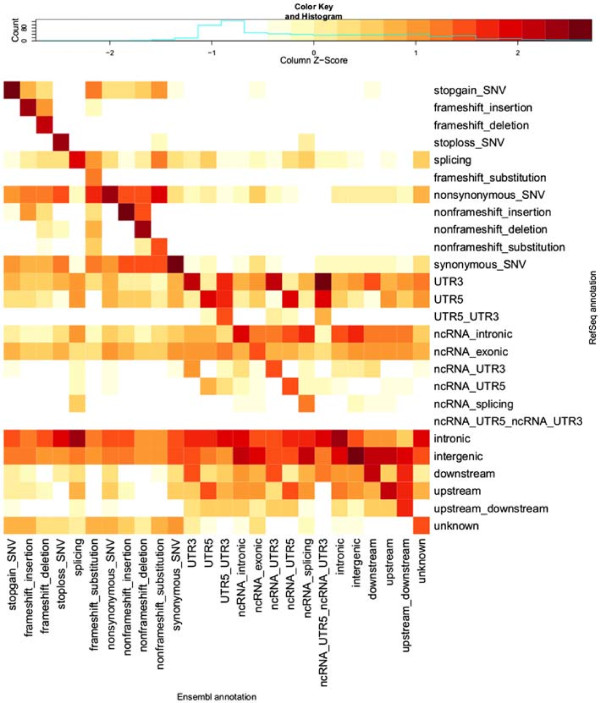
Methods: This paper quantifies the extent of differences in annotation of 80 million variants from a whole-genome sequencing study. We compare results using the RefSeq and Ensembl transcript sets as the basis for variant annotation with the software Annovar, and also compare the results from two annotation software packages, Annovar and VEP (Ensembl's Variant Effect Predictor), when using Ensembl transcripts.
Results: We found only 44% agreement in annotations for putative loss-of-function variants when using the RefSeq and Ensembl transcript sets as the basis for annotation with Annovar. The rate of matching annotations for loss-of-function and nonsynonymous variants combined was 79% and for all exonic variants it was 83%. When comparing results from Annovar and VEP using Ensembl transcripts, matching annotations were seen for only 65% of loss-of-function variants and 87% of all exonic variants, with splicing variants revealed as the category with the greatest discrepancy. Using these comparisons, we characterised the types of apparent errors made by Annovar and VEP and discuss their impact on the analysis of DNA variants in genome sequencing studies.
Conclusions: Variant annotation is not yet a solved problem. Choice of transcript set can have a large effect on the ultimate variant annotations obtained in a whole-genome sequencing study. Choice of annotation software can also have a substantial effect. The annotation step in the analysis of a genome sequencing study must therefore be considered carefully, and a conscious choice made as to which transcript set and software are used for annotation.
Figures


References
-
- Schrijver I, Aziz N, Farkas D, Furtado M, Gonzalez A, Greiner T, Grody W, Hambuch T, Kalman L, Kant J, Klein R, Leonard D, Lubin I, Mao R, Nagan N, Pratt V, Sobel M, Voelkerding K, Gibson J. Opportunities and challenges associated with clinical diagnostic genome sequencing: a report of the association for molecular pathology. J Mol Diagn. 2012;14:525–540. doi: 10.1016/j.jmoldx.2012.04.006. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
