

Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning
- PMID: 24944913
- PMCID: PMC4060303
- DOI: 10.1016/j.redox.2014.05.006
Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning
Abstract
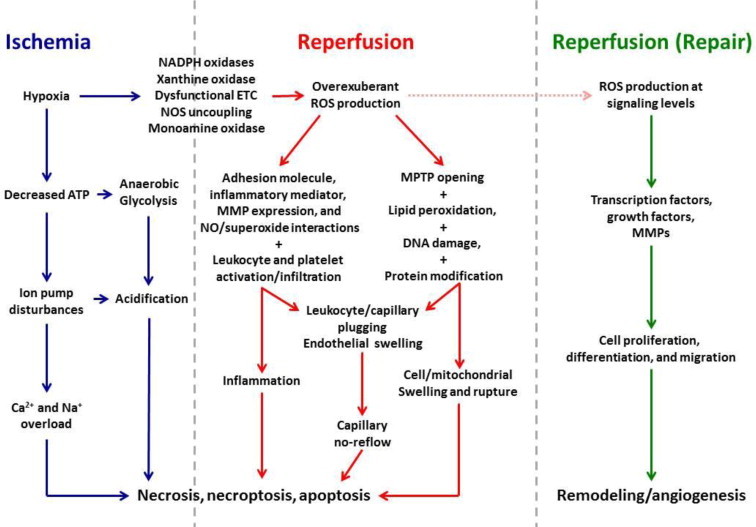
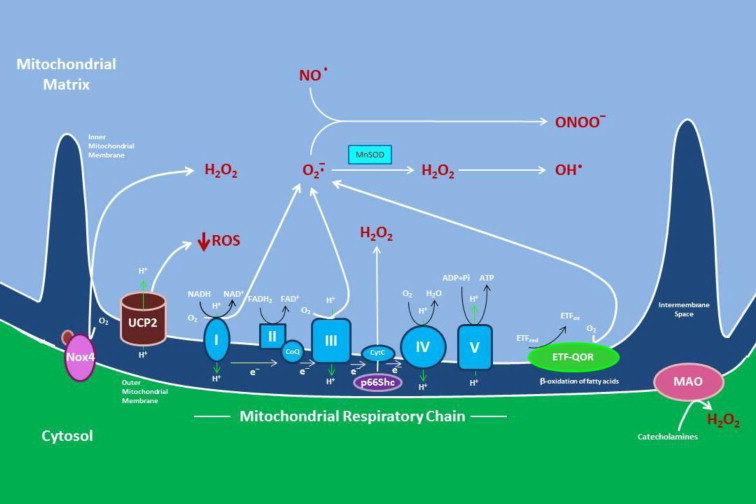
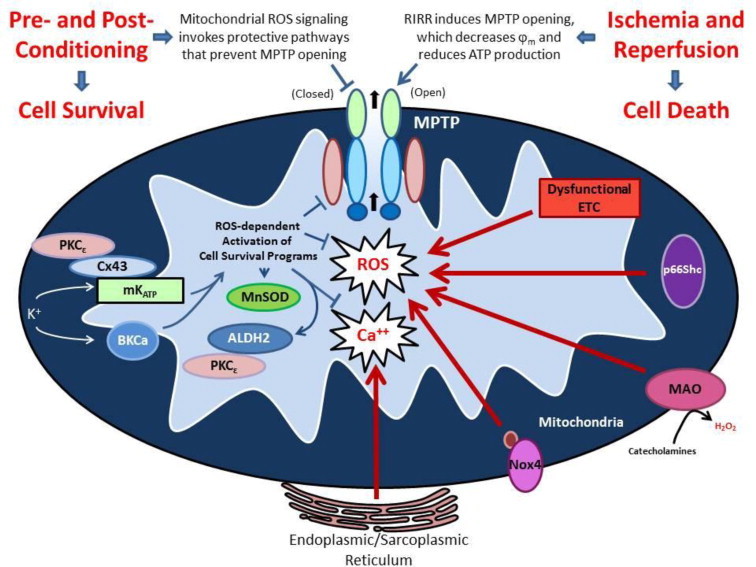
Reductions in the blood supply produce considerable injury if the duration of ischemia is prolonged. Paradoxically, restoration of perfusion to ischemic organs can exacerbate tissue damage and extend the size of an evolving infarct. Being highly metabolic organs, the heart and brain are particularly vulnerable to the deleterious effects of ischemia/reperfusion (I/R). While the pathogenetic mechanisms contributing to I/R-induced tissue injury and infarction are multifactorial, the relative importance of each contributing factor remains unclear. However, an emerging body of evidence indicates that the generation of reactive oxygen species (ROS) by mitochondria plays a critical role in damaging cellular components and initiating cell death. In this review, we summarize our current understanding of the mechanisms whereby mitochondrial ROS generation occurs in I/R and contributes to myocardial infarction and stroke. In addition, mitochondrial ROS have been shown to participate in preconditioning by several pharmacologic agents that target potassium channels (e.g., ATP-sensitive potassium (mKATP) channels or large conductance, calcium-activated potassium (mBKCa) channels) to activate cell survival programs that render tissues and organs more resistant to the deleterious effects of I/R. Finally, we review novel therapeutic approaches that selectively target mROS production to reduce postischemic tissue injury, which may prove efficacious in limiting myocardial dysfunction and infarction and abrogating neurocognitive deficits and neuronal cell death in stroke.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
