

Engineering protein farnesyltransferase for enzymatic protein labeling applications
- PMID: 24946229
- PMCID: PMC4103756
- DOI: 10.1021/bc500240p
Engineering protein farnesyltransferase for enzymatic protein labeling applications
Abstract
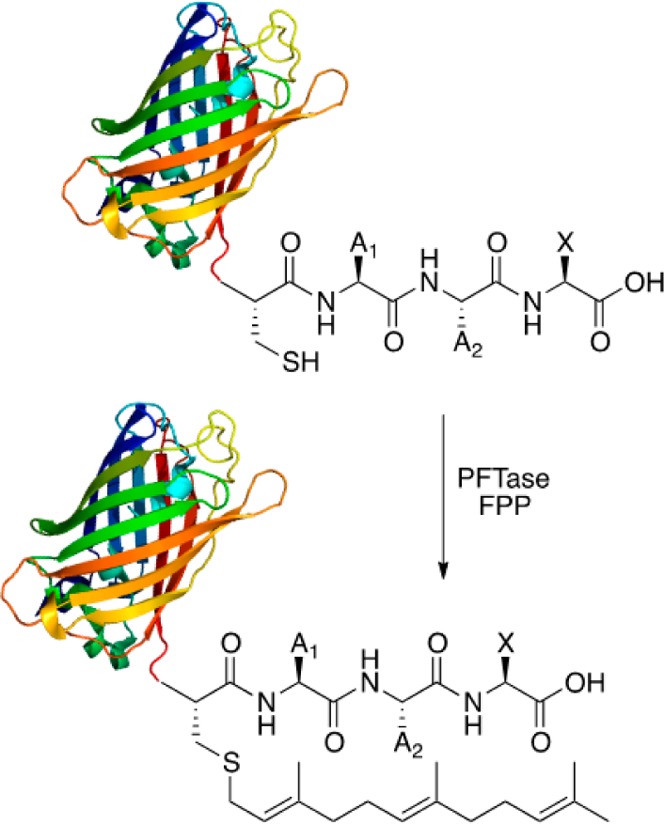
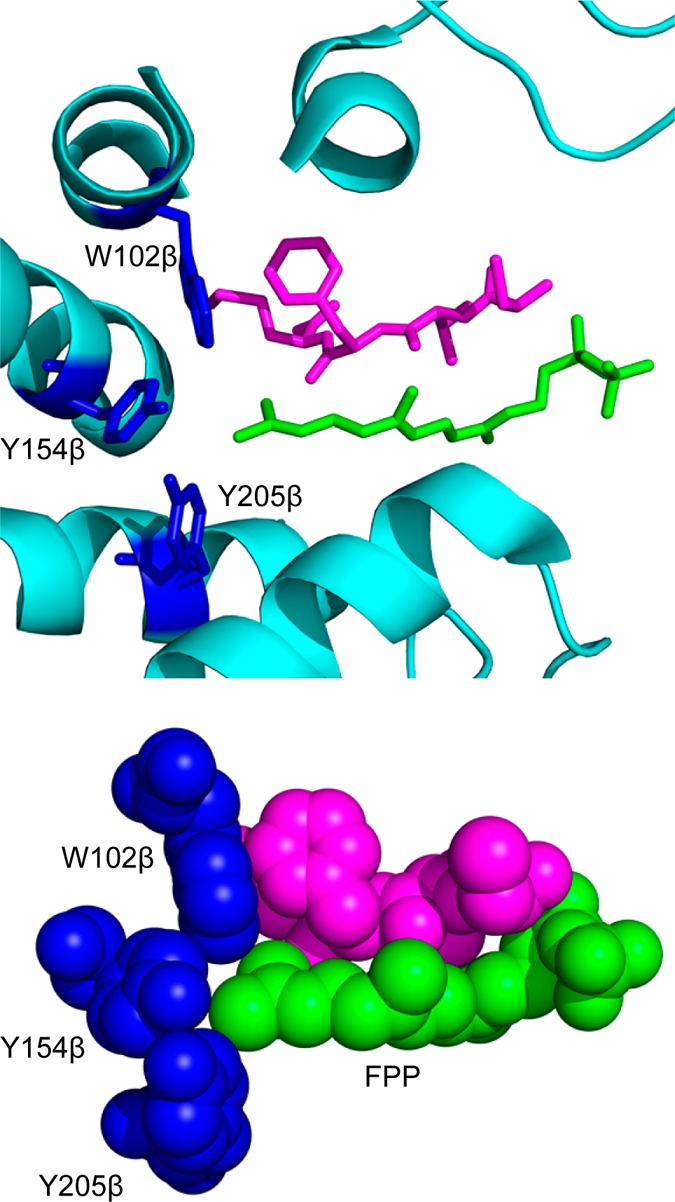
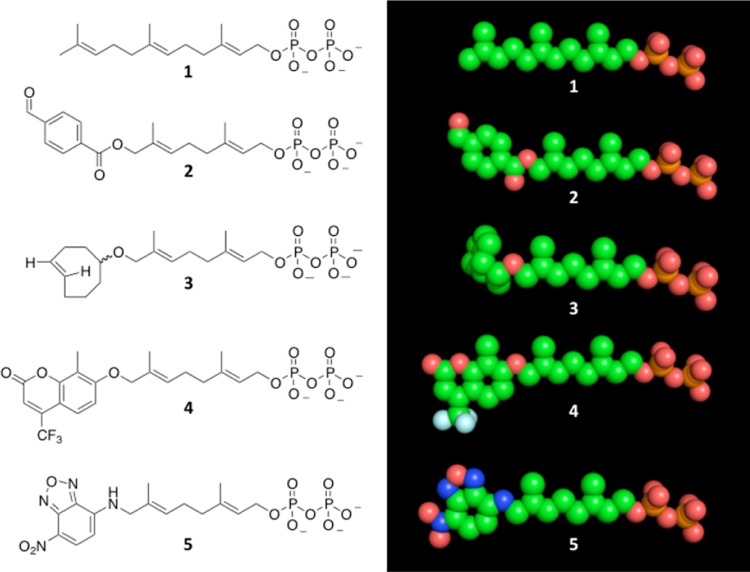
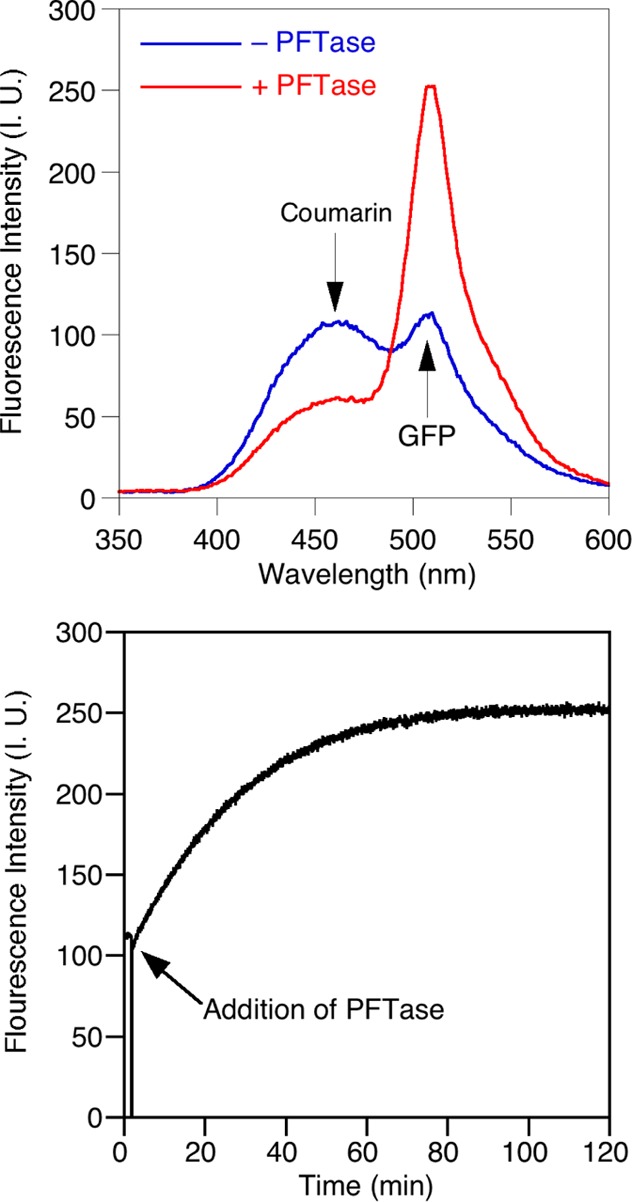
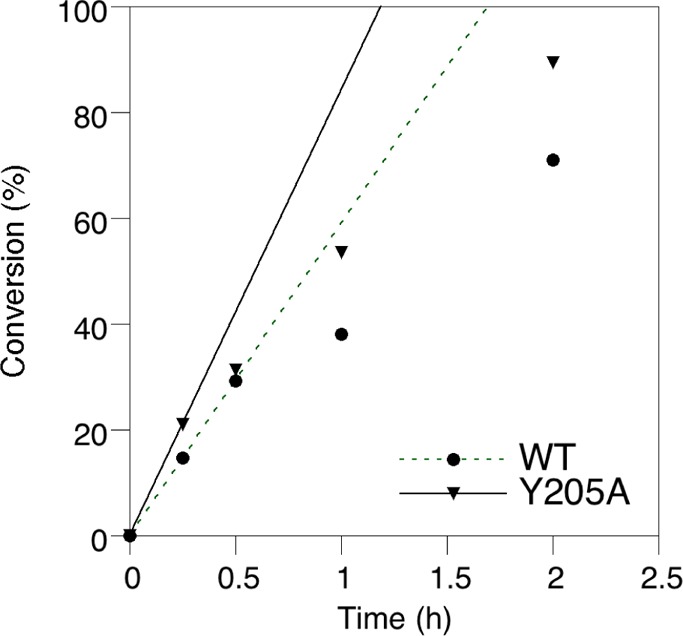
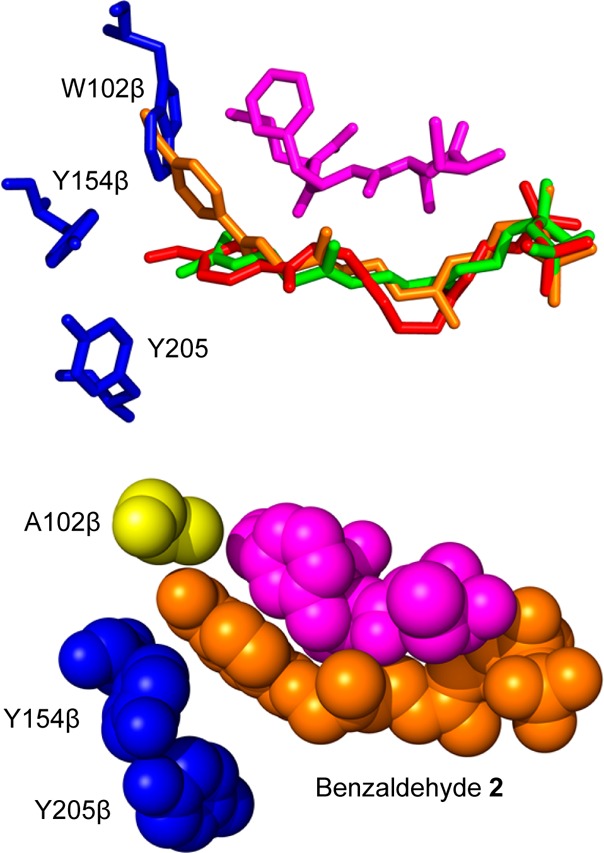
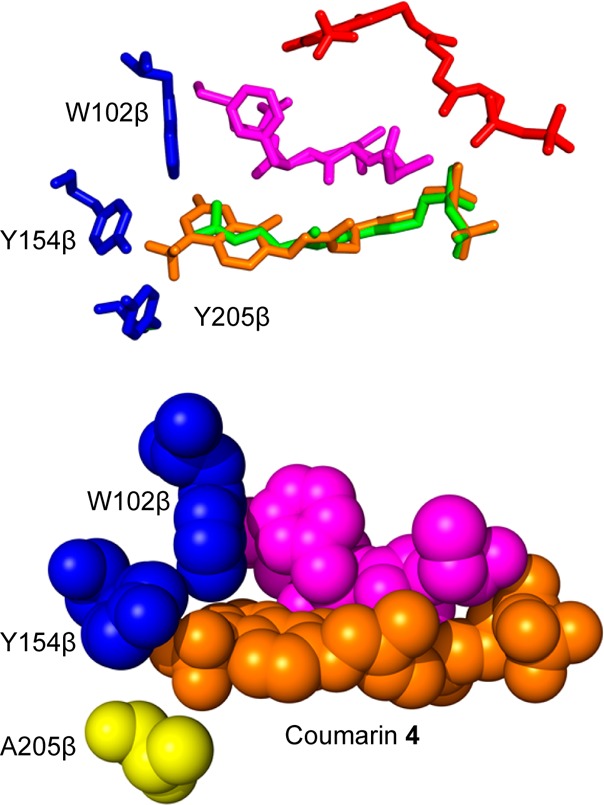
Creating covalent protein conjugates is an active area of research due to the wide range of uses for protein conjugates spanning everything from biological studies to protein therapeutics. Protein Farnesyltransferase (PFTase) has been used for the creation of site-specific protein conjugates, and a number of PFTase substrates have been developed to facilitate that work. PFTase is an effective catalyst for protein modification because it transfers Farnesyl diphosphate (FPP) analogues to protein substrates on a cysteine four residues from the C-terminus. While much work has been done to synthesize various FPP analogues, there are few reports investigating how mutations in PFTase alter the kinetics with these unnatural analogues. Herein we examined how different mutations within the PFTase active site alter the kinetics of the PFTase reaction with a series of large FPP analogues. We found that mutating either a single tryptophan or tyrosine residue to alanine results in greatly improved catalytic parameters, particularly in kcat. Mutation of tryptophan 102β to alanine caused a 4-fold increase in kcat and a 10-fold decrease in KM for a benzaldehyde-containing FPP analogue resulting in an overall 40-fold increase in catalytic efficiency. Similarly, mutation of tyrosine 205β to alanine caused a 25-fold increase in kcat and a 10-fold decrease in KM for a coumarin-containing analogue leading to a 300-fold increase in catalytic efficiency. Smaller but significant changes in catalytic parameters were also obtained for cyclo-octene- and NBD-containing FPP analogues. The latter compound was used to create a fluorescently labeled form of Ciliary Neurotrophic Factor (CNTF), a protein of therapeutic importance. Additionally, computational modeling was performed to study how the large non-natural isoprenoid analogues can fit into the active sites enlarged via mutagenesis. Overall, these results demonstrate that PFTase can be improved via mutagenesis in ways that will be useful for protein engineering and the creation of site-specific protein conjugates.
Figures
References
-
- Hackenberger C. P. R.; Schwarzer D. (2008) Chemoselective ligation and modification strategies for peptides and proteins. Angew. Chem., Int. Ed. 47, 10030–10074. - PubMed
-
- Kochendoerfer G. G. (2005) Site-specific polymer modification of therapeutic proteins. Curr. Opin. Chem. Biol. 9, 555–560. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
