

Selecting biologically informative genes in co-expression networks with a centrality score
- PMID: 24947308
- PMCID: PMC4079186
- DOI: 10.1186/1745-6150-9-12
Selecting biologically informative genes in co-expression networks with a centrality score
Abstract
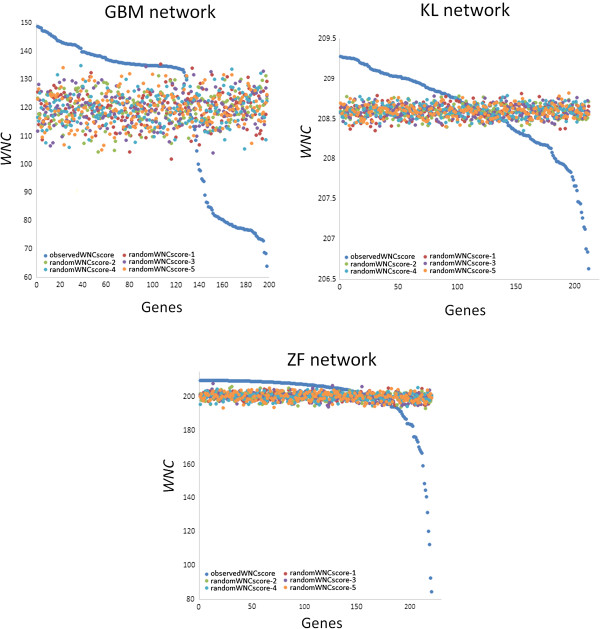
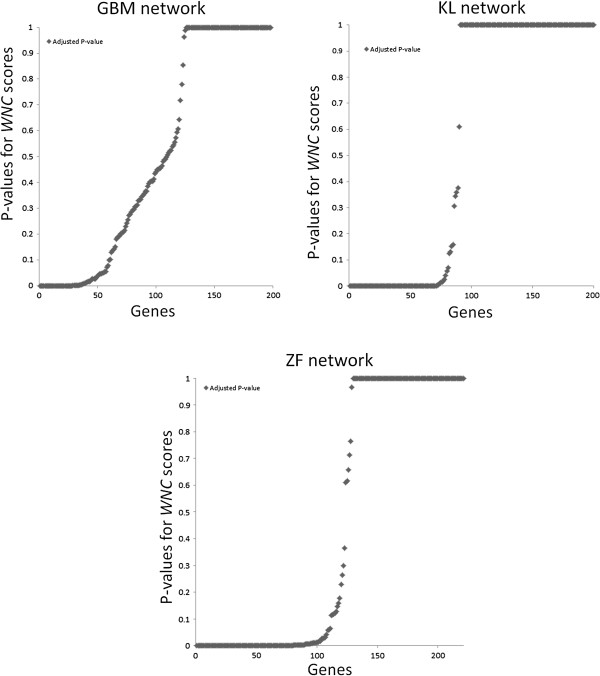
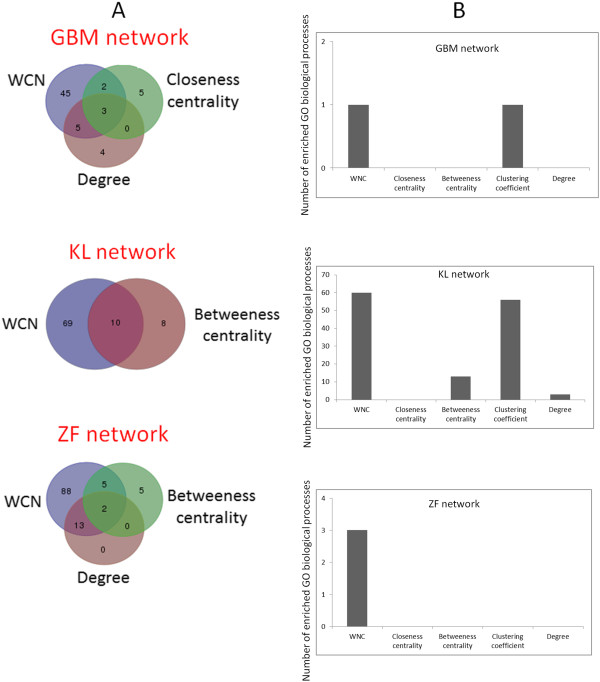
Background: Measures of node centrality in biological networks are useful to detect genes with critical functional roles. In gene co-expression networks, highly connected genes (i.e., candidate hubs) have been associated with key disease-related pathways. Although different approaches to estimating gene centrality are available, their potential biological relevance in gene co-expression networks deserves further investigation. Moreover, standard measures of gene centrality focus on binary interaction networks, which may not always be suitable in the context of co-expression networks. Here, I also investigate a method that identifies potential biologically meaningful genes based on a weighted connectivity score and indicators of statistical relevance.
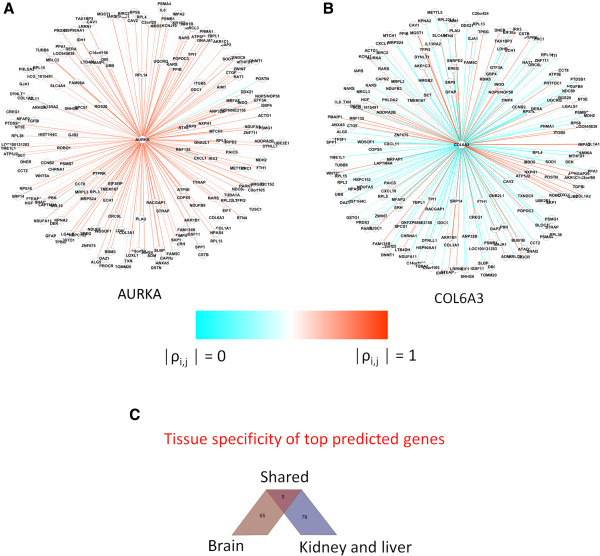
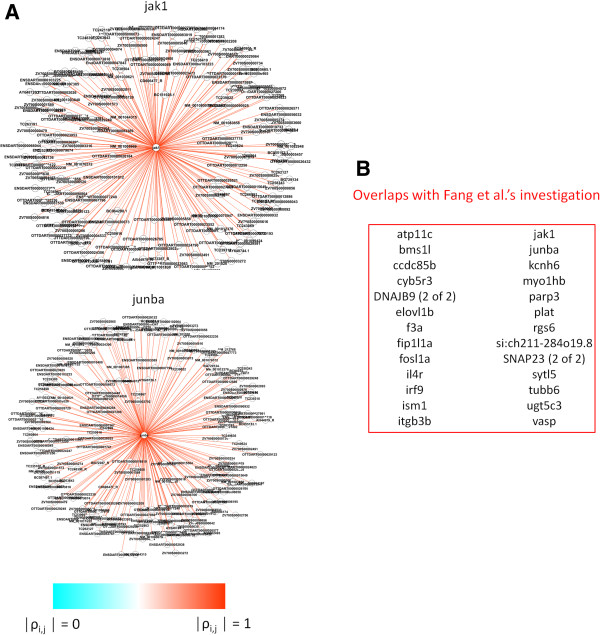
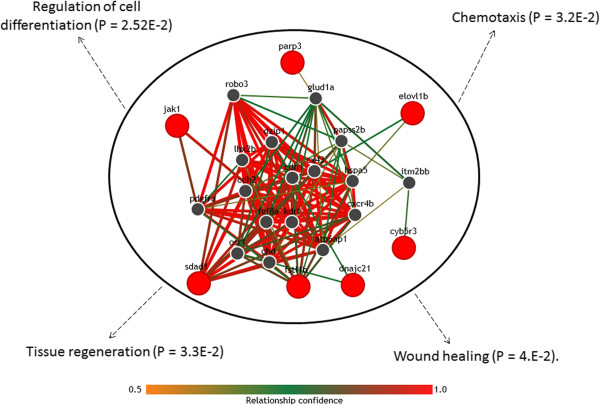
Results: The method enables a characterization of the strength and diversity of co-expression associations in the network. It outperformed standard centrality measures by highlighting more biologically informative genes in different gene co-expression networks and biological research domains. As part of the illustration of the gene selection potential of this approach, I present an application case in zebrafish heart regeneration. The proposed technique predicted genes that are significantly implicated in cellular processes required for tissue regeneration after injury.
Conclusions: A method for selecting biologically informative genes from gene co-expression networks is provided, together with free open software.
Figures
References
-
- McDermott JE, Taylor RC, Yoon H, Heffron F. Bottlenecks and hubs in inferred networks are important for virulence in Salmonella typhimurium. J Comput Biol. 2009;16:169–180. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
