

The emerging era of genomic data integration for analyzing splice isoform function
- PMID: 24951248
- PMCID: PMC4112133
- DOI: 10.1016/j.tig.2014.05.005
The emerging era of genomic data integration for analyzing splice isoform function
Abstract
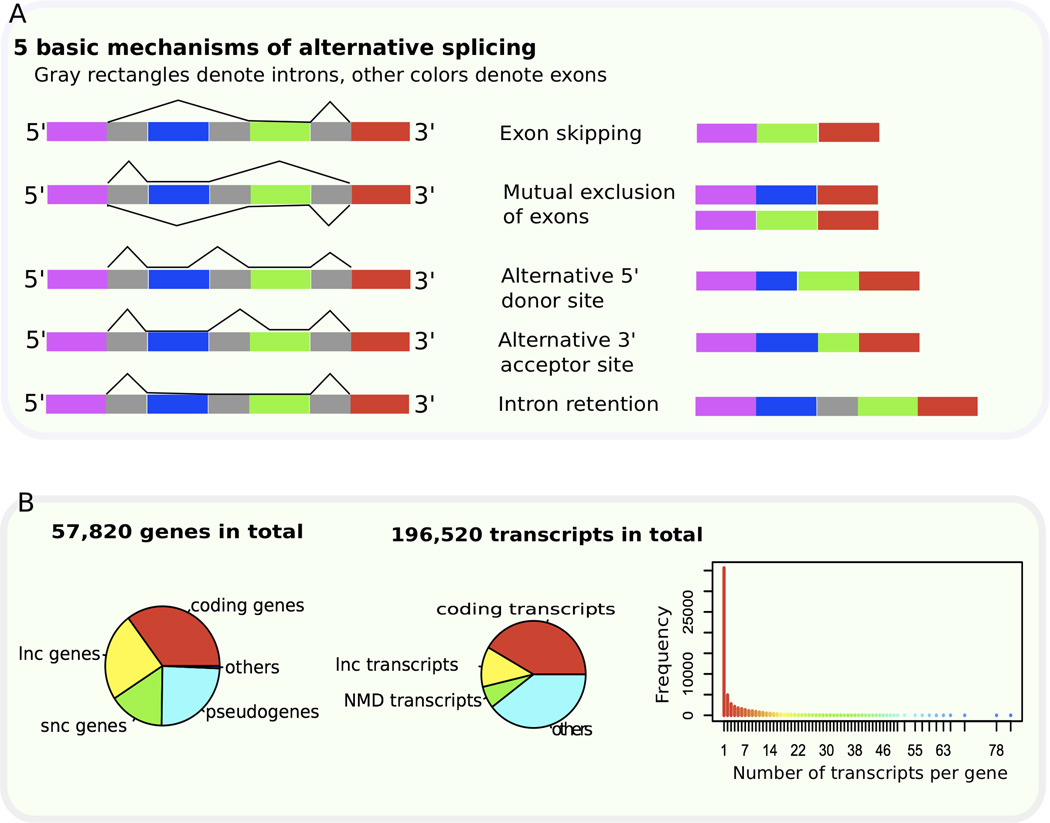
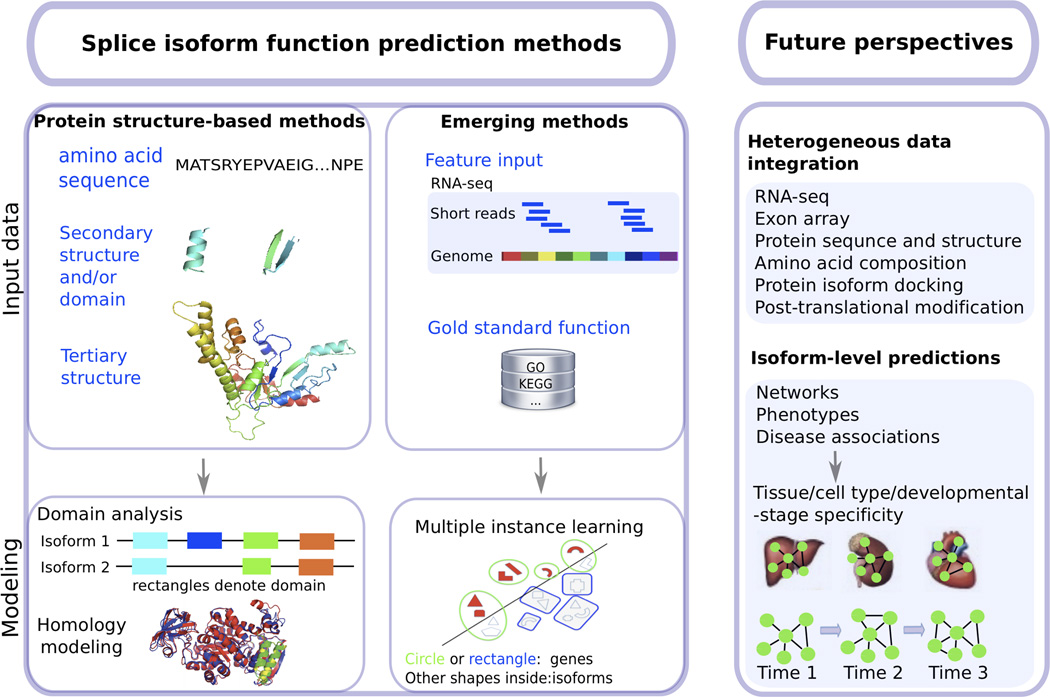
The vast majority of multi-exon genes in humans undergo alternative splicing, which greatly increases the functional diversity of protein species. Predicting functions at the isoform level is essential to further our understanding of developmental abnormalities and cancers, which frequently exhibit aberrant splicing and dysregulation of isoform expression. However, determination of isoform function is very difficult, and efforts to predict isoform function have been limited in the functional genomics field. Deep sequencing of RNA now provides an unprecedented amount of expression data at the transcript level. We describe here emerging computational approaches that integrate such large-scale whole-transcriptome sequencing (RNA-seq) data for predicting the functions of alternatively spliced isoforms, and we discuss their applications in developmental and cancer biology. We outline future directions for isoform function prediction, emphasizing the need for heterogeneous genomic data integration and tissue-specific, dynamic isoform-level network modeling, which will allow the field to realize its full potential.
Keywords: cancers; development; function prediction; genomic data integration; splice isoforms.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
