

Molecular mechanism of ligand recognition by membrane transport protein, Mhp1
- PMID: 24952894
- PMCID: PMC4195764
- DOI: 10.15252/embj.201387557
Molecular mechanism of ligand recognition by membrane transport protein, Mhp1
Abstract
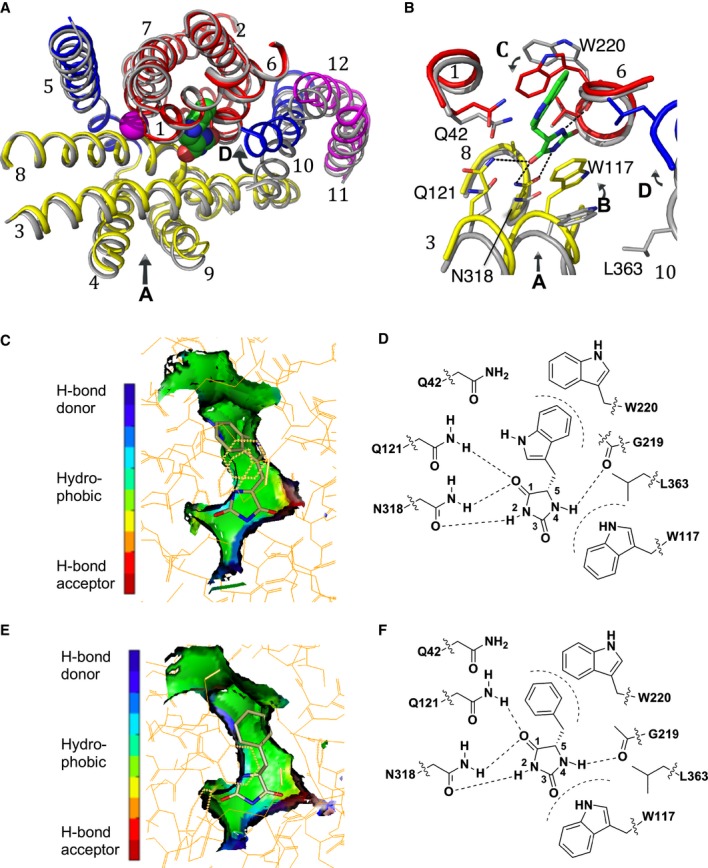
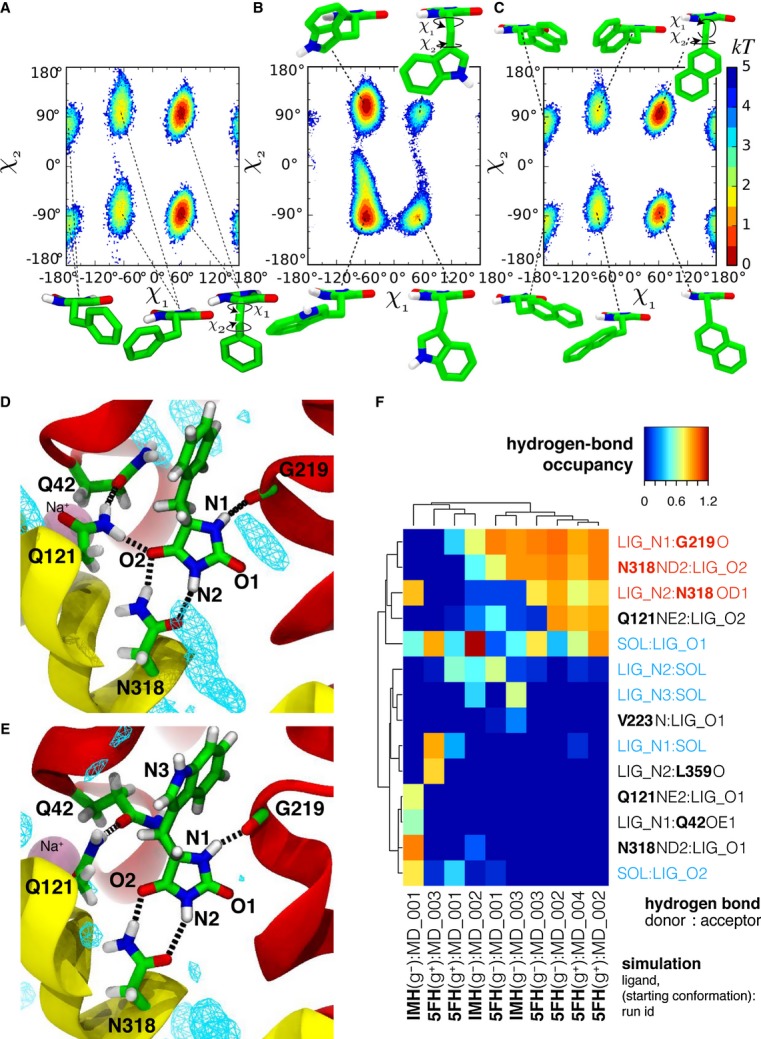
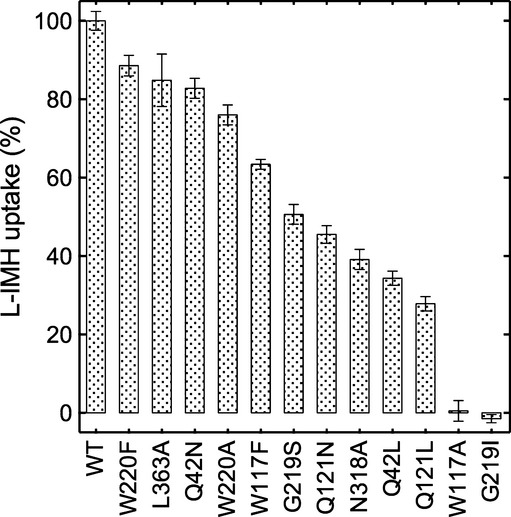
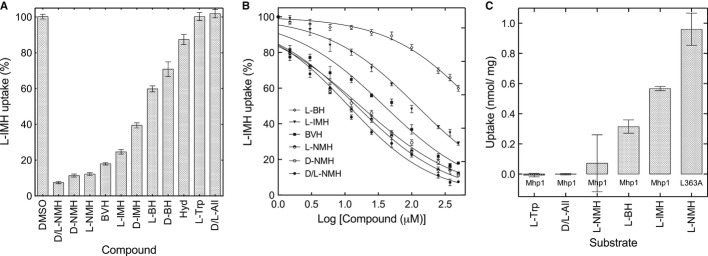
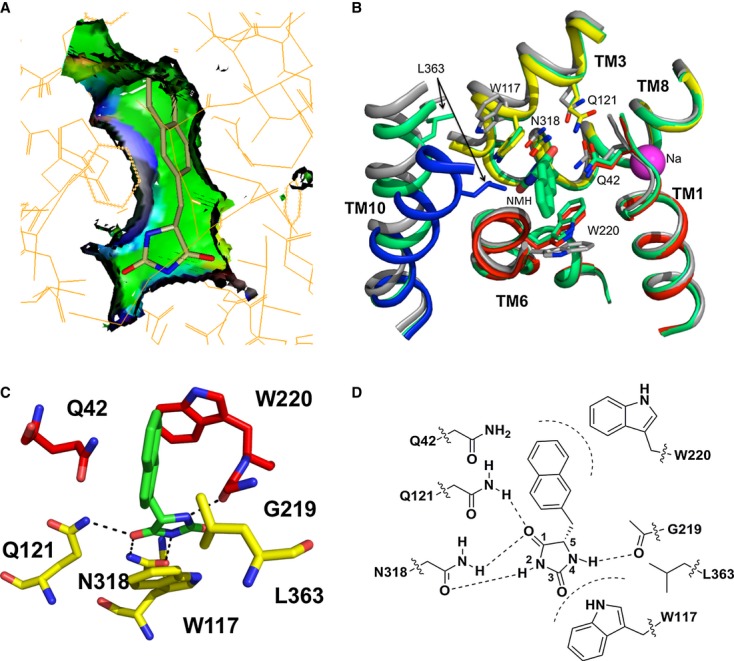
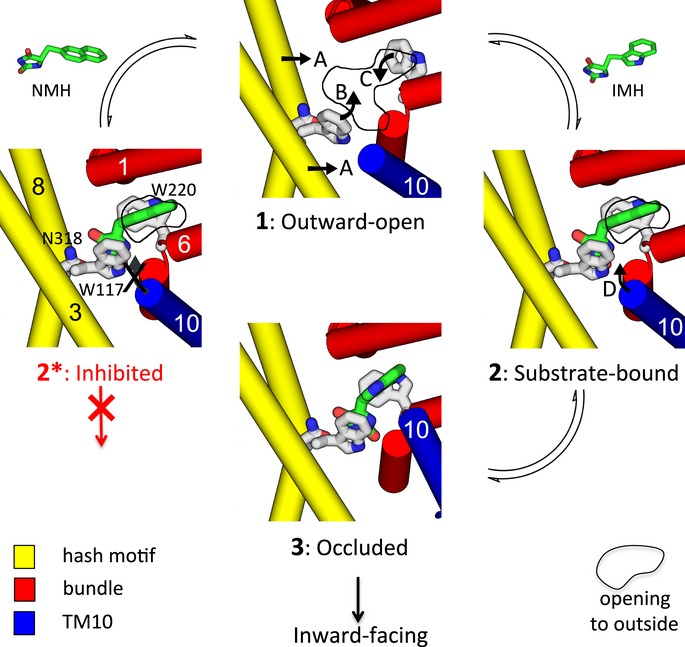
The hydantoin transporter Mhp1 is a sodium-coupled secondary active transport protein of the nucleobase-cation-symport family and a member of the widespread 5-helix inverted repeat superfamily of transporters. The structure of Mhp1 was previously solved in three different conformations providing insight into the molecular basis of the alternating access mechanism. Here, we elucidate detailed events of substrate binding, through a combination of crystallography, molecular dynamics, site-directed mutagenesis, biochemical/biophysical assays, and the design and synthesis of novel ligands. We show precisely where 5-substituted hydantoin substrates bind in an extended configuration at the interface of the bundle and hash domains. They are recognised through hydrogen bonds to the hydantoin moiety and the complementarity of the 5-substituent for a hydrophobic pocket in the protein. Furthermore, we describe a novel structure of an intermediate state of the protein with the external thin gate locked open by an inhibitor, 5-(2-naphthylmethyl)-L-hydantoin, which becomes a substrate when leucine 363 is changed to an alanine. We deduce the molecular events that underlie acquisition and transport of a ligand by Mhp1.
Keywords: Mhp1; five helix inverted repeat superfamily; hydantoin; membrane transport; molecular recognition; nucleobase‐cation‐symport, NCS1, family.
© 2014 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
Similar articles
-
Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1.Science. 2010 Apr 23;328(5977):470-3. doi: 10.1126/science.1186303. Science. 2010. PMID: 20413494 Free PMC article.
-
Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter.Science. 2008 Oct 31;322(5902):709-13. doi: 10.1126/science.1164440. Epub 2008 Oct 16. Science. 2008. PMID: 18927357 Free PMC article.
-
Conformational cycle and ion-coupling mechanism of the Na+/hydantoin transporter Mhp1.Proc Natl Acad Sci U S A. 2014 Oct 14;111(41):14752-7. doi: 10.1073/pnas.1410431111. Epub 2014 Sep 29. Proc Natl Acad Sci U S A. 2014. PMID: 25267652 Free PMC article.
-
Recent developments in nucleobase cation symporter-1 (NCS1) family transport proteins from bacteria, archaea, fungi and plants.J Biosci. 2018 Sep;43(4):797-815. J Biosci. 2018. PMID: 30207323 Review.
-
Alternating access mechanisms of LeuT-fold transporters: trailblazing towards the promised energy landscapes.Curr Opin Struct Biol. 2017 Aug;45:100-108. doi: 10.1016/j.sbi.2016.12.006. Epub 2016 Dec 30. Curr Opin Struct Biol. 2017. PMID: 28040635 Free PMC article. Review.
Cited by
-
Tales of tails in transporters.Open Biol. 2019 Jun 28;9(6):190083. doi: 10.1098/rsob.190083. Epub 2019 Jun 19. Open Biol. 2019. PMID: 31213137 Free PMC article. Review.
-
Mapping of Ion and Substrate Binding Sites in Human Sodium Iodide Symporter (hNIS).J Chem Inf Model. 2020 Mar 23;60(3):1652-1665. doi: 10.1021/acs.jcim.9b01114. Epub 2020 Mar 12. J Chem Inf Model. 2020. PMID: 32134653 Free PMC article.
-
Yeast α-arrestin Art2 is the key regulator of ubiquitylation-dependent endocytosis of plasma membrane vitamin B1 transporters.PLoS Biol. 2019 Oct 28;17(10):e3000512. doi: 10.1371/journal.pbio.3000512. eCollection 2019 Oct. PLoS Biol. 2019. PMID: 31658248 Free PMC article.
-
The Role of NCS1 in Immunotherapy and Prognosis of Human Cancer.Biomedicines. 2023 Oct 12;11(10):2765. doi: 10.3390/biomedicines11102765. Biomedicines. 2023. PMID: 37893139 Free PMC article.
-
Transmembrane helices 5 and 12 control transport dynamics, substrate affinity, and specificity in the elevator-type UapA transporter.Genetics. 2022 Aug 30;222(1):iyac107. doi: 10.1093/genetics/iyac107. Genetics. 2022. PMID: 35894659 Free PMC article.
References
-
- Altenbuchner J, Siemann-Herzberg M, Syldatk C. Hydantoinases and related enzymes as biocatalysts for the synthesis of unnatural chiral amino acids. Curr Opin Biotechnol. 2001;12:559–563. - PubMed
-
- Bommarius AS, Schwarm M, Drauz K. Biocatalysis to amino acid-based chiral pharmaceuticals - examples and perspectives. J Mol Catalysis B-Enzymatic. 1998;5:1–11.
-
- Broer S, Palacin M. The role of amino acid transporters in inherited and acquired diseases. Biochem J. 2011;436:193–211. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
- BB/L002558/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/C51725/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/B/16011/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- B19456/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/G020043/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/H000267/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/I019855/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BEP17032/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- WT089809/WT_/Wellcome Trust/United Kingdom
- BB/G023425/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- BB/C51725X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
